
Abstract
Background:
Visceral hypersensitivity is a prevalent and debilitating symptom in inflammatory bowel disease (IBD), severely impairing quality of life. Although mechanisms remain incompletely understood, recent studies have reported abnormal copper metabolism and elevated serum copper in IBD patients. However, whether copper homeostasis contributes to visceral hypersensitivity remains unknown.
Methods:
We established a dextran sulfate sodium (DSS)-induced colitis model in C57BL/6J mice to induce visceral hypersensitivity. Copper content in the terminal colon and dorsal root ganglia (DRG) was measured using a colorimetric copper assay kit. Western blot analysis evaluated expression of central sensitization markers, calmodulin-dependent kinase II (CaMKII), phosphorylated CaMKII (p-CaMKII), cyclic adenosine monophosphate response element-binding protein (CREB), and phosphorylated CREB (p-CREB) in the spinal cord and DRG. The expression of cuproptosis-related proteins, including dihydrolipoamide S-acetyltransferase (DLAT), lipoic acid synthetase (LIAS), and ferredoxin 1 (FDX1), was also measured in the terminal colon and DRG. We examined the effects of the copper chelator ammonium tetrathiomolybdate (ATTM, 10 mg/kg, i.p., daily). Expression of copper transport ATPase copper transporting alpha (ATP7A) and copper transporter 1 (CTR1) was also assessed.
Results:
DSS-induced colitis resulted in visceral hypersensitivity and central sensitization, accompanied by copper accumulation in the terminal colon and DRG, along with downregulation of DLAT, LIAS, and FDX1. ATTM reduced copper accumulation, restored cuproptosis-related protein expression, and attenuated visceral hypersensitivity and central sensitization. Notably, ATTM normalized the expression of the copper efflux transporter ATP7A and the influx transporter CTR1 in the terminal colon and DRG.
Conclusion:
Our findings for the first time definitely identify and mechanistically elucidate that copper accumulation in the terminal colon and DRG contributes to visceral hypersensitivity in DSS-induced colitis. Dysregulation of the copper transporters ATP7A and CTR1 underlies this copper dyshomeostasis. Our results uncover copper ion homeostasis as a completely unrecognized, novel mechanistic axis driving visceral hypersensitivity in IBD.
Introduction
Visceral hypersensitivity is a prevalent and debilitating manifestation of inflammatory bowel disease (IBD), characterized by exaggerated pain perception in the gastrointestinal tract in response to physiological or mildly nociceptive stimuli. 1 This chronic abdominal pain often persists despite mucosal healing and demonstrates poor correlation with the degree of intestinal inflammation, suggesting a complex neuroimmunopathological mechanism independent of active disease activity. 2 Epidemiological studies indicate that up to 60% of IBD patients report abdominal pain during their disease course, with symptoms frequently persisting even during clinical remission. 3 This condition substantially impairs patients’ quality of life and imposes a considerable socioeconomic burden on healthcare systems. 4
Visceral hypersensitivity develops and is sustained through the synergistic actions of peripheral and central sensitization. In the periphery, persistently activated immune cells (e.g. T cells and macrophages) release mediators such as proteases, histamine, serotonin, and prostaglandins. 5 These substances directly target receptors, including protease-activated receptor 2, histamine H1 receptor, and serotonin type 3 receptor on primary afferent nerve endings in the intestinal wall, lowering activation thresholds and facilitating nociceptor sensitization. 6 At the central level, persistent nociceptive input induces maladaptive changes in spinal and supraspinal circuits. This process involves microglial and astrocytic activation in the dorsal horn, leading to the release of pronociceptive factors such as brain-derived neurotrophic factor and IL-1β, which collectively enhance neuronal excitability. 7 Concurrently, impaired function of descending pain modulatory systems further reduces spinal inhibition of nociceptive signaling, thereby amplifying central sensitization. 8 Despite these advances, the precise molecular and cellular mechanisms driving visceral hypersensitivity remain incompletely understood.
Dysfunction of copper homeostasis is implicated in various neurodegenerative and metabolic diseases. Perturbations in copper metabolism can result in either deficiency or overload, contributing to conditions such as Wilson’s and Menkes’ disease. 9 In Alzheimer’s disease, elevated copper content in amyloid plaques induces oxidative stress, promotes abnormal tau phosphorylation, and enhances Aβ deposition, thereby accelerating disease progression. 10 Mutations in ATPase copper transporting alpha (ATP7A) cause intracellular copper accumulation, impairing motor neuron function and viability and thereby contributing to amyotrophic lateral sclerosis. 11 A prospective analysis of micronutrient status in quiescent IBD patients reported abnormal copper metabolism and significantly elevated serum copper content, which positively correlate with disease severity. 12 However, the role of copper homeostasis in pain regulation and visceral hypersensitivity remains to be elucidated. 13
Accordingly, we established a dextran sulfate sodium (DSS)-induced colitis model in C57BL/6J mice to induce visceral hypersensitivity. Copper content and expression of cuproptosis-related proteins, including dihydrolipoamide S-acetyltransferase (DLAT), lipoic acid synthetase (LIAS), and ferredoxin 1 (FDX1), were measured in the terminal colon and dorsal root ganglia (DRG). We further examined the effects of a copper chelator on these molecular alterations and visceral hypersensitivity in DSS-treated mice. Finally, the expression of the copper transporters ATP7A and copper transporter 1 (CTR1) was assessed in the terminal colon and DRG to elucidate potential mechanisms underlying copper dyshomeostasis in colitis.
Materials and methods
Experimental animals
All experimental procedures involving animals were approved by the Institutional Animal Care and Use Committee of Nanjing Medical University (IACUC-2412055). Adult male C57BL/6J mice (20–25 g; Qinglongshan Animal Center, Nanjing, China) were housed under specific pathogen-free conditions, with 5–6 animals per cage, in a temperature-controlled environment (22 ± 2℃) under a 12-h light/dark cycle (lights on at 8:00 AM). Food and water were provided ad libitum. To minimize stress-related effects, mice were acclimated to the testing environment for 2 days prior to behavioral experiments, which were conducted during the light phase (9:00 AM–5:00 PM). A total of approximately 50 mice were used in this study.
Establishment of the colitis mouse model and treatment with copper chelator ammonium tetrathiomolybdate (ATTM)
Colitis was induced by providing mice with drinking water containing 2% (w/v) DSS (Yeasen, 60316ES) ad libitum for seven consecutive days; fresh DSS solution was prepared daily. A 2% DSS concentration was chosen based on a well-established protocol that reliably induces acute colitis with visceral hypersensitivity in C57BL/6J mice without causing excessive mortality.14,15 Control mice received sterile drinking water for the same duration. To investigate the role of ion dyshomeostasis in colitis-associated visceral hypersensitivity, DSS-treated mice were concurrently treated with the copper chelator ATTM (MCE, HY-W076067). ATTM was freshly dissolved in sterile water each day and administered via intraperitoneal injection at a dose of 10 mg/kg throughout the 7-day induction period. This dosage was selected based on prior preclinical studies demonstrating significant neuroprotective efficacy at 10 mg/kg.16,17 Furthermore, this dose effectively reduces copper accumulation without inducing significant toxicity, rendering it suitable for repeated administration in our 7-day colitis protocol. Mice in different experimental groups were matched for age and body weight.
Record of body weight and disease activity index (DAI) score
During the experiment, general health and colitis-related symptoms were monitored daily, including body weight, stool characteristics (such as consistency and the presence of loose stools), and rectal bleeding (including occult or visible bleeding). Disease severity was assessed using the DAI, 18 calculated as: DAI = (body weight change score + fecal morphology score + rectal bleeding score)/3. Each component was scored from 0 to 4, with higher scores indicating more severe inflammation. The scoring criteria were as follows. Body weight change score: 0 (weight loss < 1%), 1 (1%–5%), 2 (5%–10%), 3 (10%–15%), and 4 (>15%); stool consistency score: 0 (normal stool), 1 (semi-solid stool), 2 (diarrhea), and 3 (no stool); and rectal bleeding score: 0 (none), 1 (mild), 2 (moderate), 3 (severe), and 4 (massive bleeding).
Assessment of visceral hypersensitivity
Visceral hypersensitivity was evaluated using two methods. First, visceral pain thresholds were measured via colorectal distension. 15 Briefly, under 2% isoflurane anesthesia, mice were secured on a transparent restraining platform. A PE-60 catheter fitted with a polyethylene balloon (1.5 cm × 1 cm) was inserted transanally, positioning the balloon 0.5 cm from the anal verge, and the catheter was secured to the tail. After a 10-min recovery and acclimation period, the balloon was inflated at a constant rate of 3 mmHg/s until a visceromotor response (abdominal lifting or body arching) was observed. The pressure at which this response occurred was recorded as the pain threshold. Each mouse was tested three times, and the average value was calculated. All behavioral testing was performed in a group-blinded manner to minimize bias.
Additionally, we utilized a step-down avoidance assay in response to focal colorectal dilation (fCRD) to assess visceral hypersensitivity.19,20 To facilitate acclimation, mice were housed in the experimental room for 3 days prior to testing. All procedures were performed in a quiet, dimly lit environment to minimize external disturbances. Following anesthesia with 2% isoflurane, a latex balloon catheter lubricated with glycerol was gently inserted 0.5 cm into the rectum and secured to the tail base with tape. During a 3-day adaptation period, the catheter remained in situ while mice were placed in an inverted transparent cylinder (diameter 15 cm, height 1.5 cm) for 10 min daily. Formal testing commenced on day 4. Each mouse was placed on a 1.5 cm platform within the transparent cylinder. Upon stepping down onto a cardboard surface (40 cm × 40 cm), the balloon was inflated to 0, 10, or 30 mmHg for 30 s. Due to anticipated visceral pain from CRD, mice might hesitate to step down. Each mouse underwent six trials at each pressure level, with 5-min intervals between trials. Latency (seconds) from cylinder removal to when all four paws contacted the cardboard (maximum 120 s) was recorded as a measure of visceral hypersensitivity.
Measurement of colon length and histopathological scoring of terminal colonic tissue
Following behavioral testing, mice were deeply anesthetized with 5% isoflurane, and colon length was measured. Colitis severity was assessed via hematoxylin and eosin (HE) staining of terminal colon sections.21,22 Briefly, tissues were fixed in 10% formaldehyde, dehydrated through a graded ethanol series, paraffin embedded, and sectioned at 5 μm thickness. Histological scoring evaluated crypt injury and inflammation score as follows. Crypt injury score: 0 (normal crypts), 1 (mild changes), 2 (moderate changes), 3 (severe changes in crypts and loss of surface epithelial cells), and 4 (complete destruction of crypts but presence of surface epithelial cells); inflammation score: 0 (no inflammation), 1 (mild thickening of the intrinsic layer), 2 (moderate thickening of the lamina propria with infiltration of inflammatory cells), and 3 (significant thickening of the lamina propria and infiltration of many inflammatory cells).
Western blot analysis
Following colon length measurement, segments of the terminal colon, L4-5 spinal cord, and L6-S2 DRG were harvested for western blot analysis. For protein extraction, tissues were homogenized in RIPA lysis buffer supplemented with protease inhibitor cocktail for 30 min. Lysates were centrifuged at 4℃ and 2000 × g for 5 min, and the supernatant was collected. Protein concentrations were determined using a BCA protein quantification kit. Target proteins were separated by SDS-PAGE and transferred onto PVDF membranes via a semi-dry transfer method. Membranes were then blocked in a Tris-buffered saline with Tween-20 (TBST) containing 10% low-fat dry milk for 2 h at room temperature. To investigate central sensitization in colitis model mice, we examined expression of key markers CaMKII, phospho-CaMKII, CREB, and phospho-CREB in the spinal cord and DRG. PVDF membranes were incubated overnight at 4℃ with primary antibodies against CaMKII (rabbit monoclonal anti-CaMKII, Abclonal, A0198, 1:500), phospho-CaMKII (rabbit polyclonal anti-Phospho-CaMKII, CST, 12818, 1:1000), CREB (rabbit monoclonal anti-CREB, Abclonal, A10826, 1:500), and phospho-CREB (rabbit polyclonal anti-Phospho-CREB, Abclonal, AP0019, 1:500). Following 5-min washes with TBST at room temperature three times, membranes were incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (H+L; Abcam, ab6721, 1:3000) for 2 h at room temperature. Rabbit monoclonal anti-β-actin (Proteintech, 66009-1-Ig, 1:5000) was used as a loading control. Protein bands were visualized using enhanced chemiluminescence reagent and imaged with a Molecular Imager. Band intensities were quantified using ImageJ software.
Additionally, expression of cuproptosis-related proteins DLAT (rabbit polyclonal anti-DLAT, Abmart, PS08581, 1:500), LIAS (rabbit polyclonal anti-LIAS, Abmart, PH2927, 1:500), and FDX1 (rabbit polyclonal anti-FDX1, Abmart, T510671, 1:500) was examined in the terminal colon and DRG of colitis model mice. The expression of the aforementioned proteins was also assessed in colitis model mice following treatment with ATTM. To investigate mechanisms underlying copper dyshomeostasis, expression of copper transporters ATP7A (rabbit monoclonal anti-ATP7A, Abcam, ab308524, 1:500) and CTR1 (rabbit polyclonal anti-CTR1/SLC31A1, Abmart, T510261, 1:500) was also evaluated in the terminal colon and DRG.
Colorimetric copper quantitative detection
To investigate the role of copper homeostasis in pain regulation and visceral hypersensitivity, copper content was measured in the terminal colon and DRG using a copper assay kit (Solarbio, BC5755) according to the manufacturer’s protocol. In brief, approximately 1 cm of terminal colon and 10–30 mg of L6-S2 DRG were harvested, snap-frozen in liquid nitrogen, and homogenized on ice in ice-cold distilled water at a tissue-to-water ratio of 1:5 to 1:10 (w/v). Homogenates were centrifuged at 10,000 × g for 10 min at 4°C. For the assay, 10 μL of supernatant was added to each sample well, followed by sequential addition of 150 μL Reagent 1 and 50 μL of Reagent 2. After thorough mixing, the plate was incubated at 37°C for 5 min, and absorbance was measured at 580 nm using a microplate reader. Copper concentrations were calculated from a copper sulfate standard curve.
Statistical analysis
Statistical analyses were performed using GraphPad Prism software (version 9.0). All quantitative data were presented as mean ± standard error of the mean (mean ± SEM). Comparisons between two groups were analyzed by unpaired t-test, while multiple group comparisons were conducted using one-way or two-way analysis of variance as appropriate. A p-value of less than 0.05 was considered statistically significant.
Results
DSS treatment induced acute colitis in mice
We induced colitis in mice by oral administration of 2% DSS for seven consecutive days (Figure 1(a)). During DSS treatment, mice progressively exhibited colitis symptoms, including deteriorated mental state, decreased activity, reduced food and water intake, diarrhea, and fecal blood. Body weight decreased from day 4, while the DAI increased from day 3 in mice after DSS treatment (Figure 1(b) and (c)).

DSS treatment induced acute colitis in mice. (a) Scheme of the model establishment of DSS-induced colitis; (b) DSS treatment significantly decreased the body weight of mice; (c) DSS treatment significantly increased DAI scores; (d) DSS treatment significantly decreased visceral pain threshold in response to colorectal distension; (e) Scheme of the step-down avoidance test; (f) Control mice exhibited significant avoidance at 30 but not 0 or 10 mmHg fCRD; (g) DSS-treated mice displayed marked aversive behavior at 10 mmHg, with significantly shorter latency to step down from the platform during trials 3–6.
Visceral pain thresholds, assessed via colorectal distension, were significantly reduced from day 3 after DSS treatment (Figure 1(d)). In the step-down avoidance test (Figure 1(e)), control mice showed no aversion to 10 mmHg fCRD, like sham stimulation (0 mmHg), but exhibited significant avoidance at 30 mmHg (Figure 1(f)). Conversely, DSS-treated mice displayed marked aversive behavior even at 10 mmHg, with significantly shorter latency to step down from the platform during trials 3–6 (Figure 1(g)). These results demonstrated DSS treatment induced deteriorated health status and visceral hypersensitivity in mice.
DSS aggravated colonic histopathology and up-regulated expression of central sensitization markers
Following behavioral testing, we harvested the colon and found the colon was significantly shorter after DSS treatment (6.160 ± 0.4904 vs 9.300 ± 0.5099 cm; Figure 2(a)). Additionally, these mice exhibited loose stools, edema, and colon congestion. Histopathological analysis of the terminal colon revealed severe mucosal damage, characterized by inflammatory cell infiltration, crypt distortion, and disordered glandular architecture, resulting in significantly higher histopathological scores (Figure 2(b)). DSS treatment also up-regulated the expression of central sensitization markers p-CaMKII and p-CREB in both the spinal cord and DRG (Figure 2(c) and (d)). Collectively, these data demonstrate that oral administration of DSS successfully established a mouse model of colitis with visceral hypersensitivity.

DSS aggravated colonic histopathology and up-regulated expression of central sensitization markers. (a) DSS treatment significantly shortened the colon length of mice. Data are presented as mean ± SEM and compared with unpaired t-test. N = 10 mice per group; (b) DSS treatment significantly increased histopathological scores in the terminal colon of mice. Images are in 100× magnification. Data are presented as mean ± SEM and compared with unpaired t-test. N = 4 mice per group; (c–d) Western blot shows that DSS treatment significantly up-regulated the expression of central sensitization markers p-CaMKII and p-CREB in the spinal cord and DRG of mice.
Copper homeostasis imbalance was observed in DSS-induced colitis model mice
To explore the contribution of copper homeostasis to pain regulation and visceral hypersensitivity, we quantified copper content in the terminal colon and DRG of DSS-induced colitis mice. Colorimetric assays revealed a significant increase in copper ions in both tissues following DSS treatment (Figure 3(a) and (c)). In parallel with this copper accumulation, western blot analysis showed a pronounced downregulation of the cuproptosis-related proteins DLAT, LIAS, and FDX1 in the terminal colon and DRG (Figure 3(b) and (d)). This reduction likely represents a compensatory response aimed at limiting the availability of lipoylated copper-binding targets and thereby mitigating cuproptotic stress. Nevertheless, the persistently elevated copper content suggests that this protective adaptation is insufficient to prevent copper overload-induced neuronal hypersensitivity. Collectively, these findings indicate that copper homeostasis is significantly disrupted in this colitis model.

Copper homeostasis imbalance was observed in DSS-induced colitis model mice. (a/c) DSS treatment significantly increased copper ion content in the terminal colon and DRG of mice. Data are presented as mean ± SEM and compared with unpaired t-test. N = 4 mice per group; (b/d) Western blot shows that DSS treatment significantly down-regulates key cuproptosis-related proteins DLAT, LIAS, and FDX1 in the terminal colon and DRG of mice.
ATTM attenuated copper accumulation and restored cuproptosis-related proteins expression in DSS-induced colitis mice
To investigate the etiological role of copper accumulation in visceral hypersensitivity in this colitis model, we treated DSS-induced colitis mice with intraperitoneal injection of ATTM, a highly specific copper chelator (Figure 4(a)). As shown, ATTM treatment attenuated copper accumulation (Figure 4(b)) and restored expression of cuproptosis-related proteins DLAT, LIAS, and FDX1 expression in the terminal colon and DRG (Figure 4(c) and (d)).

ATTM attenuated copper accumulation and restored cuproptosis-related protein expression in DSS-induced colitis mice. (a) Scheme diagram of ATTM treatment protocol in DSS-induced colitis mice; (b) ATTM treatment significantly decreased copper ion content in both the terminal colon and DRG of DSS-induced colitis mice. Data are presented as mean ± SEM and compared with unpaired t-test. N = 4 mice per group; (c–d) Western blot shows that ATTM treatment significantly elevated key cuproptosis-related proteins DLAT, LIAS, and FDX1 in the terminal colon and DRG of DSS-induced colitis mice.
ATTM alleviated visceral hypersensitivity and central sensitization in DSS-induced colitis mice
ATTM treatment attenuated copper accumulation and restored cuproptosis-related protein expression in DSS-induced colitis mice. However, macroscopic signs of colitis, including colon shortening, weight loss, and elevated DAI scores, showed no significant improvement in the ATTM-treated mice compared to the colitis mice (Figure 5(a)–(c)). Notably, ATTM elevated the visceral pain threshold in response to colorectal distension and shortened the latency period under fCRD at 10 mmHg in DSS-induced colitis mice (Figure 5(d) and (e)). Furthermore, ATTM significantly downregulated the expression of central sensitization markers, p-CaMKII and p-CREB, in the spinal cord and DRG (Figure 5(f) and (g)). These results collectively indicate that copper chelation can specifically alleviate visceral hypersensitivity but not the overall inflammatory pathology.
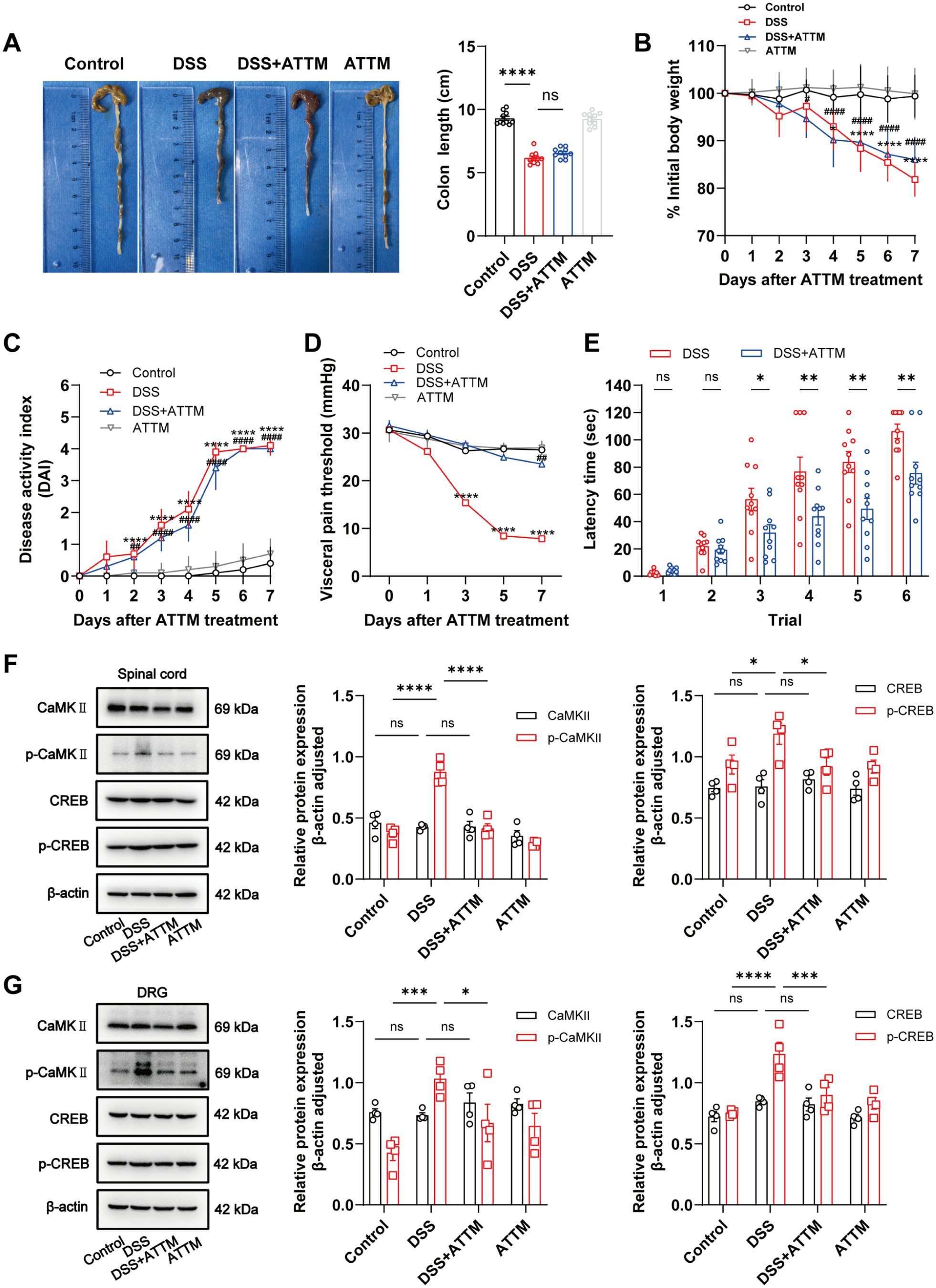
ATTM alleviated visceral hypersensitivity and central sensitization in DSS-induced colitis mice. (a) ATTM treatment showed no significant improvement in DSS-induced colon shortening. Data are presented as mean ± SEM and compared with unpaired t-test. N = 10 mice per group; (b) ATTM treatment showed no significant improvement in DSS-induced weight loss. Data are presented as mean ± SEM and compared with two-way ANOVA with Bonferroni’s multiple comparisons test. N = 10 mice per group; (c) ATTM treatment showed no significant improvement in DSS-induced elevated DAI scores. Data are presented as mean ± SEM and compared with two-way ANOVA with Bonferroni’s multiple comparisons test. N = 10 mice per group; (d) ATTM treatment elevated visceral pain threshold in response to colorectal distension of DSS-induced colitis mice. Data are presented as mean ± SEM and compared with two-way ANOVA with Bonferroni’s multiple comparisons test. N = 10 mice per group; (e) ATTM treatment shortened the latency time under fCRD at 10 mmHg in DSS-induced colitis mice. Data are presented as mean ± SEM and compared with two-way ANOVA with Bonferroni’s multiple comparisons test. N = 10 mice per group; (f–g) Western blot shows that ATTM treatment significantly down-regulated the expression of central sensitization markers p-CaMKII and p-CREB in the spinal cord and DRG of DSS-induced colitis mice.
ATTM reversed copper transporters ATP7A and CTR1 expression in the terminal colon and DRG
Although copper chelation specifically attenuated copper accumulation, restored cuproptosis-related protein expression, and alleviated visceral hypersensitivity in colitis mice, the underlying mechanism remains unclear. Intracellular copper homeostasis is highly dependent on various copper transporters, including copper efflux transporters, including ATP7A and ATPase copper transporting beta polypeptide (ATP7B), as well as copper influx transporters, such as CTR1. As ATP7A is primarily responsible for copper efflux in the periphery, like the intestine, whereas ATP7B functions predominantly in the liver, we accordingly assessed the expression of the copper efflux transporter ATP7A and copper influx transporter CTR1 in DSS-induced colitis mice. As shown, in both the terminal colon and DRG, DSS treatment significantly increased ATP7A expression while decreasing CTR1 expression (Figure 6(a) and (b)). Notably, ATTM reversed these alterations (Figure 6(c) and (d)). These findings suggest that dysregulated copper transporter expression may contribute to copper accumulation in the colon and DRG, leading to copper homeostasis imbalance and visceral hypersensitivity.
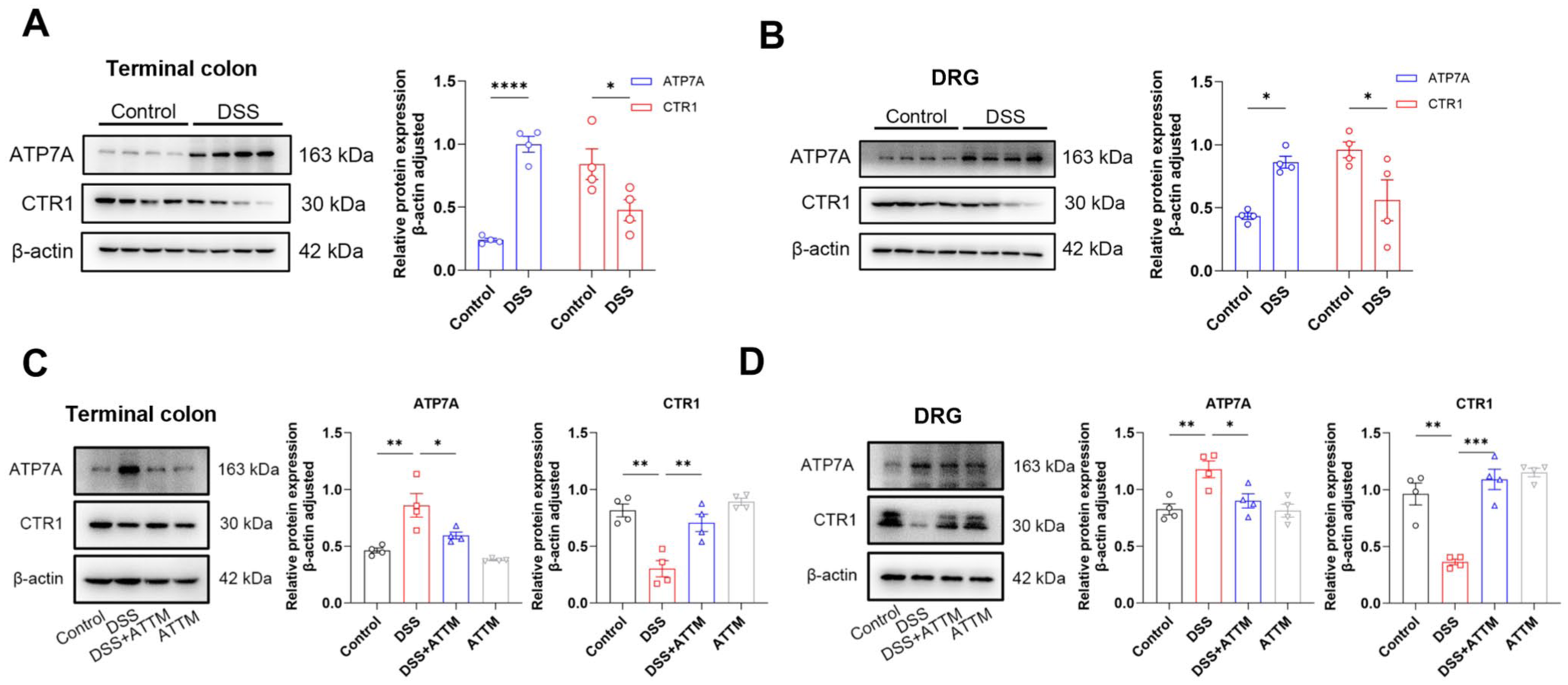
ATTM reversed copper transporters ATP7A and CTR1 expression in the terminal colon and DRG. (a–b) Western blot shows that DSS treatment significantly increased the copper efflux transporter ATP7A expression while decreasing copper influx transporter CTR1 expression in the terminal colon and DRG of mice. (c–d) Western blot shows that ATTM treatment significantly decreased ATP7A expression while increasing CTR1 expression in the terminal colon and DRG of DSS-induced colitis mice.
Discussion
In this study, DSS-induced colitis mice presented visceral hypersensitivity and central sensitization, which were associated with significant copper accumulation in the terminal colon and DRG and concurrent downregulation of key cuproptosis-related proteins – DLAT, LIAS, and FDX1. Copper chelator ATTM mitigated copper accumulation, restored cuproptosis-related protein expression, and attenuated both visceral hypersensitivity and central sensitization. Importantly, ATTM reversed the dysregulation of copper transporters in DSS-induced colitis, reducing ATP7A expression while restoring CTR1 level in the terminal colon and DRG. Collectively, our results uncover copper ion homeostasis as a completely unrecognized, novel mechanistic axis driving visceral hypersensitivity in IBD.
Chronic abdominal pain is one of the most prevalent and debilitating symptoms in patients with IBD, 23 typically presenting as gradual, diffuse, and persistent discomfort with heightened sensitivity to mechanical stretch and distension stimuli.24,25 This constellation of symptom poses formidable therapeutic challenges, compromising patients’ quality of life and imposing a considerable socioeconomic burden. Current pharmacological approaches – including 5-aminosalicylic acid drugs (mesalazine), 26 glucocorticoids (prednisone), 27 or immunomodulators (azathioprine) 28 – demonstrate only limited analgesic efficacy and are associated with serious adverse events such as ischemic colitis and cardiovascular ischemia. 29 Accordingly, an in-depth investigation into alternative pathophysiological mechanisms underlying IBD-associated visceral hypersensitivity is clearly warranted.
The European Society of Clinical Nutrition and Metabolism recommends regular micronutrient monitoring in IBD patients. 30 Based on a prospective analysis in patients with quiescent IBD, serum analyses reveal decreased zinc and iron concentrations, elevated copper, and an increased copper/zinc ratio compared to healthy subjects. These perturbations worsen with disease progression, as zinc and iron decline, while the copper/zinc ratio increases. 12 Similarly, serum element traces showed decreased serum iron, zinc, and selenium and increased copper and manganese in pediatric IBD. 31 Consistent with these clinical observations, we detected significant copper accumulation in both the terminal colon and DRG. While copper dyshomeostasis is implicated in diverse pathological conditions – including Wilson’s disease, Alzheimer’s disease, Huntington’s disease, and various malignancies 32 – its specific contribution to visceral hypersensitivity remains to be elucidated. As an essential micronutrient, copper serves as a critical catalytic cofactor for fundamental biological processes, including mitochondrial respiration, antioxidant defense, and biocompound synthesis. Under physical conditions, copper is homeostatically maintained at low micromolar concentrations; however, even modest elevations can induce cytotoxicity and trigger cell death pathways. 33
Programmed cell death, encompassing apoptosis, ferroptosis, necroptosis, pyroptosis, and autophagy, is fundamentally involved in IBD pathogenesis. 34 Although apoptosis is frequently associated with neuronal loss in chronic pain, it plays a relatively indirect and largely secondary role to upstream inflammatory signals in the acute sensitization of nociceptive circuits, as seen in our colitis model. 35 Ferroptosis, driven by iron-dependent lipid peroxidation, contributes to neuropathic pain and inflammatory pain primarily via glial activation and enhanced oxidative stress.36,37 Although both cuproptosis and ferroptosis are metal-dependent and impair mitochondrial integrity, cuproptosis is uniquely triggered by copper binding to lipoylated tricarboxylic acid cycle proteins, 38 causing direct mitochondrial proteotoxic stress, in contrast to the membrane lipid damage characteristic of ferroptosis. Necroptosis, mediated by the RIPK1/RIPK3/MLKL axis, promotes robust inflammatory cytokine release and sustains inflammation in colitis models, 39 but its direct role in sensory neuron sensitization remains poorly defined. Metal ion dyshomeostasis can disrupt neuronal excitability and synaptic function through this pathway, often independently of extensive inflammatory cascades or overt cell death. This key distinction indicates that cuproptosis may mediate pain specifically via bioenergetic deficits and impaired signal transduction in sensory neurons, thus representing a mechanistically distinct and potentially complementary therapeutic target to apoptosis, ferroptosis, or necroptosis.
In our study, we observed downregulation of DLAT – a key lipoylated target protein – in both the terminal colon and DRG. Acylation-dependent DLAT aggregation only disrupts pyruvate metabolism, suppresses ATP production, and arrests mitochondrial energy metabolism.40,41 We also detected downregulation of the Fe-S cluster proteins FDX1 and LIAS. FDX1 functions as a critical upstream regulator of cuproptosis, 42 promoting copper-induced cell death by sustaining protein lipoylation and maintaining copper in its active reduced form. 43 LIAS is the primary lipoic acid synthesis, essential for pyruvate dehydrogenase complex activity and directly modulating the copper-binding capacity of lipoylated proteins; its dysfunction may compromise cellular tolerance to copper toxicity. 44
ATTM chelates copper ions in the gastrointestinal tract and bloodstream to form inert, unabsorbed copper thiomolybdate complexes, thereby blocking systemic copper absorption. 45 In our study, ATTM not only attenuated copper accumulation and restored the expression of cuproptosis-related proteins but also reinstated physiological lipoylation and copper-chelating capacity by normalizing DLAT, LIAS, and FDX1 levels. This shift from cuproptotic stress to neuronal alleviated both visceral hypersensitivity and central sensitization. Collectively, these results establish copper accumulation as a critical driver of visceral hypersensitivity in experimental colitis.
Cellular copper homeostasis is tightly controlled by specific transporters: the copper efflux transporters ATP7A and ATP7B (ATPase copper transporting beta), and the copper importer CTR1. ATP7A mainly mediates copper export in peripheral tissues such as the intestine, whereas ATP7B primarily governs hepatic copper metabolism, we investigated the expression of the efflux transporter ATP7A and influx transporter CTR1 in a DSS-induced colitis model. Under physiological conditions, ATP7A promotes copper ion incorporation into cuproenzymes in the trans-Golgi network; upon copper overload, it translocates from the Golgi to the plasma membrane to export excess copper. 46 Conversely, CTR1 – a high-affinity copper importer localized at the plasma membrane – mediates specific uptake of extracellular copper. 47 Our results showed that DSS treatment significantly upregulated ATP7A expression and downregulated CTR1 expression in both the terminal colon and DRG, and these changes were effectively reversed by ATTM administration.
In conclusion, the pivotal innovation of our study lies in the first identification of copper ion homeostasis as a completely unrecognized, novel mechanistic axis driving visceral hypersensitivity in IBD, breaking the long-standing research focus on a single neuroimmune-inflammatory framework in this field. Further, copper-related biomarkers such as DLAT and FDX1 can help facilitate patient stratification. Furthermore, we pioneer the validation that the copper chelator ATTM can specifically reverse copper homeostasis disorder and alleviate visceral hypersensitivity, providing a brand-new copper-targeted therapeutic strategy for intractable visceral pain in IBD. Copper-targeted drugs hold substantial translational potential for IBD. Clinical trials confirm the safety of agents like copper ionophore disulfiram and copper chelator ATTM, while preclinical studies validate their efficacy in reversing mucosal inflammation. 48 Copper-targeted therapies for IBD offer distinct advantages: they modulate inflammatory pathways like NF-κB via COMMD1 to suppress excessive immune responses, maintain intestinal barrier integrity by regulating HIF-1α, and balance oxidative stress by scavenging reactive oxygen species or mitigating copper-induced damage. They also improve gut immune homeostasis and microbiota balance, synergize with antioxidants or immunomodulators, and exhibit favorable biosafety, providing personalized and effective options for disease control.48,49
However, this study has several limitations. First, due to technical and equipment limitations, we did not incorporate the abdominal wall electromyography, which is an objective physiological data in the assessment of visceral pain threshold. Second, the specific cell type involved in copper accumulation and cuproptosis within the terminal colon and DRG, as well as their molecular targets, was not validated in this study. Third, no in vitro cell experiments or clinical sample validations were conducted to confirm the association between copper homeostasis and visceral hypersensitivity in human IBD patients. Additionally, the expression patterns of copper transporters (ATP7A, CTR1) in human diseases were not verified, reducing the clinical reference value of the conclusions.
Footnotes
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.*
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by grants from the Nanjing Medical Science and Technology Development Fund (ZKX23040) and the National Natural Science Foundation of China (82400516).