
Abstract
The downregulation of mu-opioid receptor (MOR) expression in dorsal root ganglia (DRG) following peripheral nerve injury plays a pivotal role in neuropathic pain development and contributes to the reduced efficacy of morphine analgesia. The molecular mechanisms underlying this transcriptional silencing remain poorly understood. In this study, we demonstrate that peripheral nerve injury induces upregulation of methyl-CpG-binding domain protein 1 (MBD1) and histone lysine methyltransferase SUV39H1, leading to increased trimethylation of histone H3 at lysine 9 (H3K9me3) at the Oprm1 gene locus in injured DRG neurons. Genetic ablation of MBD1 reversed injury-induced MOR downregulation at both mRNA and protein levels, attenuated H3K9me3 enrichment at the Oprm1 promoter, and alleviated neuropathic pain behaviors, despite persistent SUV39H1 upregulation. Mechanistic studies revealed that nerve injury enhances the recruitment of SUV39H1 and H3K9me3 to the Oprm1 promoter, which is dependent on MBD1. Our findings establish a novel epigenetic mechanism wherein MBD1-mediated recruitment of SUV39H1 promotes H3K9me3-dependent transcriptional silencing of Oprm1 in DRG neurons following peripheral nerve injury.
Keywords
Strengths and limitations of this study
This study represents, to our knowledge, the first comprehensive investigation elucidating the epigenetic mechanism by which SUV39H1 and MBD1 coordinately silence MOR expression in injured DRG neurons under neuropathic pain conditions following peripheral nerve injury.
Our findings provide novel insights into the molecular interplay between DNA methylation and histone modifications in regulating MOR expression and their contribution to neuropathic pain pathogenesis.
However, it should be noted that the current research focuses exclusively on DRG mechanisms and may not fully represent epigenetic regulatory processes in other neural structures involved in neuropathic pain.
Introduction
Neuropathic pain resulting from nerve injury represents a challenging chronic condition characterized by diverse symptoms including spontaneous pain, mechanical allodynia, and thermal hypersensitivity. 1 Current therapeutic strategies, including opioid analgesics and nonsteroidal anti-inflammatory drugs, often prove inadequate due to their limited specificity for the underlying mechanisms of neuropathic pain.2,3 Among these treatments, opioid drugs such as morphine and fentanyl exert their analgesic effects primarily through activation of the mu-opioid receptor (MOR), encoded by the Oprm1 gene, and are widely used for managing moderate to severe acute and chronic pain.4,5 However, the clinical efficacy of opioid analgesics is significantly diminished in neuropathic pain conditions, a phenomenon consistently observed in both clinical settings and animal models.6,7 This reduced efficacy has been partially attributed to the downregulation of MOR expression at both transcriptional and translational levels in injured dorsal root ganglion (DRG) neurons following peripheral nerve injury.8,9 Therefore, elucidating the mechanisms underlying nerve injury-induced MOR downregulation in DRG neurons may provide novel therapeutic targets for improved neuropathic pain management.
Epigenetic regulation through histone modification represents a crucial mechanism controlling gene expression. This process involves the transfer of methyl groups to specific amino acid residues on histone proteins, leading to chromatin structural changes and subsequent modulation of gene expression. 10 Suppressor of variegation 3-9 homolog 1 (SUV39H1), a histone lysine methyltransferase, specifically catalyzes the formation of trimethylated histone H3 at lysine 9 (H3K9me3), resulting in chromatin condensation and transcriptional repression.11,12 Our previous work demonstrated that inhibition of nerve injury-induced SUV39H1 upregulation in DRG and spinal cord not only alleviates neuropathic pain symptoms but also reverses MOR downregulation in injured DRG neurons. 13 Nevertheless, the precise molecular mechanisms by which SUV39H1 contributes to MOR downregulation remain to be fully elucidated.
DNA methylation represents another critical epigenetic mechanism that regulates gene expression through multiple pathways, including the recruitment of transcriptional repressors such as methyl-CpG-binding domain (MBD) proteins. 14 MBD family proteins play pivotal roles in determining transcriptional states and mediating crosstalk between DNA methylation and histone modifications. 15 Among these proteins, MBD1 has been shown to specifically recognize methylated DNA and interact with SUV39H1, suggesting its involvement in epigenetic gene silencing.16,17 Notably, increased methylation of the MOR gene promoter has been observed in injured DRG following peripheral nerve injury. 18 Building upon our previous findings that both SUV39H1 and MBD1 are crucial regulators in neuropathic pain, we hypothesize that SUV39H1 may regulate MOR expression in injured DRG through MBD1-mediated histone modification.
In the present study, we demonstrate that peripheral nerve injury induces SUV39H1 upregulation and subsequent H3K9me3 formation in a MBD1-dependent manner. This epigenetic mechanism contributes to nerve injury-induced MOR downregulation in DRG neurons, ultimately leading to the development and maintenance of chronic neuropathic pain. Our findings provide novel insights into the molecular mechanisms by which SUV39H1 and MBD1 coordinately regulate MOR expression through epigenetic modifications in neuropathic pain conditions.
Materials and methods
Animal
The Mbd1-/- mice used in this study were generously provided by Dr. Yuan-Xiang Tao (Rutgers State University of New Jersey) and maintained on a C57BL/6J genetic background through complete backcrossing. 19 These mice were bred by crossing heterozygous pairs in our facility. Previous studies have demonstrated that Mbd1-/- mice exhibit normal viability, physical appearance, and locomotor activity in both genders.19,20 For our experiments, we utilized adult male Mbd1-/- mice and their wild-type littermates (25–30 g body weight). All animals were maintained under standard housing conditions with a 12-h light/dark cycle and provided with ad libitum access to food and water.
All experimental procedures were conducted in strict accordance with the ethical guidelines established by the International Association for the Study of Pain and were approved by the Institutional Animal Care and Use Committee at Zhujiang Hospital, Southern Medical University. We implemented stringent measures to minimize animal usage and reduce potential discomfort throughout the study. Additionally, all experimental procedures were performed by investigators blinded to the treatment conditions to ensure objective data collection.
Neuropathic pain model
The mouse model of neuropathic pain was established through unilateral L4 spinal nerve ligation (SNL) following our previously established protocol.20–22 Briefly, mice were anesthetized using isoflurane, followed by a 10-mm longitudinal incision in the lumbar region to expose the L4 transverse process. The process was carefully removed to visualize the corresponding spinal nerve. The left L4 spinal nerve was isolated and tightly ligated using 7-0 silk suture, followed by complete transection distal to the ligation site. Sham-operated control animals underwent identical surgical procedures except for the nerve ligation and transection. Finally, the muscle layers and skin were sutured separately for proper wound closure.
Behavioral testing
Behavioral assessments, including mechanical, thermal, and cold sensitivity tests, were conducted following our established protocols.20,21 Prior to baseline testing, mice were acclimated to the testing environment for 1–2 h daily over 2–3 days. A minimum 1-h interval was maintained between different behavioral tests to minimize potential interference.
Paw withdrawal frequencies were evaluated using von Frey filaments as previously described.20,22 Mice were individually placed in transparent Plexiglas chambers on an elevated wire mesh platform. The plantar surface of each hind paw was stimulated with a 0.4 g von Frey filament for approximately 1 s, with 10 stimuli applied to each paw at 5-min intervals. Paw withdrawal frequency was calculated as the percentage of positive responses ([number of paw withdrawals/10 trials] × 100 = % response frequency).
Paw withdrawal latencies were assessed using a radiant heat source (Analgesic Meter, IITC Inc/Life Science Instruments, Model 336).20,21 Mice were placed in Plexiglas chambers on a glass platform, and a focused heat beam was directed to the mid-plantar surface of each hind paw. The apparatus automatically recorded paw withdrawal latency, defined as the time interval between heat onset and paw withdrawal. Each paw was tested five times with 5-min intervals, with a 20-s cutoff to prevent tissue damage.
Paw withdrawal latencies were measured using a temperature-controlled metal plate maintained at 0°C.22,23 Mice were placed in Plexiglas chambers on the cold plate, and withdrawal latency was recorded as the time from paw contact to withdrawal. Each ipsilateral hind paw was tested three times with 10-min intervals. A 20-s cutoff was implemented to prevent cold-induced tissue injury.
Plasmid constructs and virus or siRNA production
The full-length mouse Mbd1 cDNA was synthesized and amplified from DRG total RNA using gene-specific primers (Table S1), followed by cloning into a pro-viral plasmid. Recombinant adeno-associated virus serotype 5 (rAAV5) particles containing the Mbd1 expression construct (rAAV5-Mbd1) were generated at the University of North Carolina Vector Core Facility. Control viral particles expressing enhanced green fluorescent protein (rAAV5-GFP) were obtained from the same facility. These viral preparations were kindly provided by Dr. Yuan-Xiang Tao, with detailed production and characterization information available in our previously published protocols. 20 For gene silencing experiments, Suv39h1-specific siRNA (sc-38464B) and scrambled control siRNA (sc-37007) were commercially.
DRG primary neuron cell culture and siRNA or viral transfection
Primary DRG neuronal cultures were established following previously established protocols.20,22,24 Briefly, 21-day-old wild-type and Mbd1-/- mice were euthanized via isoflurane overdose, and DRGs from cervical to lumbar levels were rapidly dissected and collected in cold neurobasal medium (Gibco/Life Technologies) supplemented with 10% fetal bovine serum (Procell), 2% B-27 supplement, 1% L-glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin (Invitrogen/Thermo Fisher Scientific). Following washing and centrifugation, DRG tissues were enzymatically dissociated in Ca2+- and Mg2+-free HBSS (Life Technologies) containing 5 mg/ml dispase and 1 mg/ml collagenase type I at 37°C for 20–25 min. The dissociated cells were resuspended in complete neurobasal medium and plated onto poly-D-lysine-coated (50 µg/ml, Sigma) six-well plates at a density of 1.5–4 × 105 cells per well. Cultures were maintained in a humidified incubator at 37°C with 5% CO2.
For viral transduction, 2 µl of AAV5 viral particles (titer ≥ 1 × 1012/ml) were added to each well containing 2 ml medium after 24 h of initial plating. For siRNA transfection experiments, 100 nM siRNA was complexed with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol and applied to cultures 24 h post-plating. Following 48–72 h of viral transduction and/or siRNA transfection, cells were harvested for subsequent mRNA and protein expression analyses.
Western blotting analysis
As previously described,20,23 bilateral L4 dorsal root ganglia (DRGs) or cultured neuronal cells were harvested at specified time points post-surgery and homogenized at 4°C. Protein extraction was performed using RIPA lysis buffer (Thermo Fisher Scientific, Waltham, MA, USA), followed by quantification of protein concentration using a BCA assay kit (Solarbio, Beijing, China). Protein samples were denatured by boiling for 5 min in loading buffer and subsequently separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The separated proteins were then transferred electrophoretically onto polyvinylidene difluoride (PVDF) membranes.
Following blocking, the PVDF membranes were incubated with the following primary antibodies at 4°C overnight: rabbit anti-μ-opioid receptor (MOR; 1:500, Neuromics, Edina, MN, USA), mouse anti-methyl-CpG-binding domain protein 1 (MBD1; 1:500, kindly provided by Dr. Yuan-Xiang Tao), rabbit anti-suppressor of variegation 3-9 homolog 1 (SUV39h1; 1:1000, Cell Signaling Technology, Danvers, MA, USA), rabbit anti-trimethylated histone H3 lysine 9 (H3K9me3; 1:500, Signalway Antibody, College Park, MD, USA), rabbit anti-histone H3 (1:1000, Cell Signaling Technology), and rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1:1000, Santa Cruz Biotechnology, Dallas, TX, USA). After incubation with appropriate secondary antibodies, protein bands were visualized using chemiluminescent substrates (Thermo Scientific) and quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Protein expression levels were normalized to either histone H3 or GAPDH as internal controls.
RNA extraction and reverse transcription PCR (RT-PCR)
RNA isolation and quantitative reverse transcription PCR (qRT-PCR) were performed as previously described.20,23 Briefly, L4 dorsal root ganglia (DRGs) from mice subjected to spinal nerve ligation (SNL) surgery or primary DRG cells cultured for 48 h were collected. Total RNA was extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA), and RNA purity and concentration were determined using a NanoDrop 8000 Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA). First-strand cDNA synthesis was performed with 1 μg of total RNA using the PrimeScript™ RT Reagent Kit (TaKaRa, Dalian, China) according to the manufacturer’s instructions.
Quantitative PCR amplification was conducted using the SYBR Premix Ex Taq II kit (TaKaRa) in a 10 μl reaction volume on a BIO-RAD CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA). The thermal cycling conditions were as follows: initial denaturation at 95°C for 3 min, followed by 40 cycles of 95°C for 10 s, 60°C for 30 s, and 72°C for 30 s. Melt curve analysis was performed after amplification to verify product specificity. Relative mRNA expression levels were calculated using the comparative Ct method (2-ΔΔCt). Primer sequences were designed using Primer 3.0 software and are listed in Table S1. All data were normalized to the housekeeping gene Gapdh.
For single-cell qRT-PCR analysis, freshly dissociated DRG neurons from adult mice were plated on poly-D-lysine-coated (50 μg/ml; Sigma-Aldrich, St. Louis, MO, USA) culture dishes for 4 h. Individual DRG neurons were categorized by size: large (>35 μm), medium (25–35 μm), and small (<25 μm). Single neurons were collected in PCR tubes containing 5–10 μl of cell lysis buffer (Signosis, Santa Clara, CA, USA) as previously described. 22 Following incubation and centrifugation, the supernatant was used as a template for reverse transcription. Subsequent qRT-PCR procedures were performed using the single-cell real-time PCR assay kit (Signosis) according to the manufacturer’s protocol. All primer sequences are provided in Table S1.
Co-immunoprecipitation assay
To investigate potential protein-protein interactions between SUV39h1 and MBD1, co-immunoprecipitation (Co-IP) assays were conducted following established protocols.20,25 Dorsal root ganglia (DRGs) were dissected from 3 to 4 week-old wildtype and Mbd1-/- mice, washed twice with 300 μl ice-cold phosphate-buffered saline (PBS), and homogenized in 700 μl NP-40 lysis buffer (50 mmol/l Tris, pH 8.0; 150 mmol/l NaCl; 0.1% SDS; 1.0% NP-40; protease inhibitor cocktail; and 10 μM trichostatin A [TSA]) using a glass tissue grinder. The homogenates were sonicated and centrifuged at 12,000 × g for 15 min at 4°C.
Protein concentrations were quantified using a BCA assay, and 400 μg of total protein from each sample was allocated for immunoprecipitation, with the remainder reserved for input controls. Lysates were pre-cleared by incubation with 50 μl Protein A/G agarose beads (Santa Cruz Biotechnology, Dallas, TX, USA) for 1 h at 4°C. Subsequently, pre-cleared lysates were incubated overnight at 4°C with 8 μl undiluted SUV39h1 antiserum (Cell Signaling Technology, Danvers, MA, USA) or control IgG, followed by addition of 40 μl Protein A/G agarose beads and rotation for 4 h at 4°C.
Immunocomplexes were washed four times with NP-40 lysis buffer and eluted by boiling in 2× Laemmli sample buffer. Samples were resolved by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes for immunoblotting. Protein interactions were assessed by probing membranes with antibodies against MBD1 and SUV39h1 as described in the Western blotting methodology.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed using the EZ ChIP Kit (Millipore, Billerica, MA, USA) according to the manufacturer’s protocol with modifications as previously described.20–22 Briefly, DRG tissues from wildtype mice were homogenized and cross-linked with 1% formaldehyde for 10 min at room temperature. The cross-linking reaction was terminated by adding 0.25 M glycine. Following centrifugation at 2000 × g for 5 min at 4°C, the pellet was resuspended in SDS lysis buffer containing protease inhibitor cocktail and incubated on ice for 10 min.
Chromatin was sheared by sonication to generate DNA fragments ranging from 200 to 1000 bp, as verified by agarose gel electrophoresis. After pre-clearing with protein G agarose beads, samples were incubated overnight at 4°C with 3 μg of specific antibodies: mouse anti-SUV39h1 (Abcam, Cambridge, UK; ab12405, ChIP grade) or rabbit anti-trimethylated histone H3 lysine 9 (H3K9me3; Signalway Antibody, College Park, MD, USA). Parallel samples were incubated with 3 μg of normal IgG as negative controls. Input controls, representing 10%–20% of the total chromatin, were set aside for normalization.
Immunocomplexes were collected using protein G agarose beads, washed extensively, and eluted. Cross-links were reversed by incubation at 65°C overnight, followed by proteinase K treatment. DNA fragments were purified using spin columns and analyzed by quantitative PCR with specific primers listed in Table S1. The relative enrichment of target DNA fragments was calculated by normalizing to input controls.
Statistical analysis
For in vivo investigations, experimental mice were randomly allocated to various treatment groups using a computer-generated randomization schedule. In vitro experiments were conducted with cells uniformly suspended and randomly distributed across experimental wells using a systematic randomization protocol. All quantitative data are presented as mean ± standard error of the mean (SEM).
Statistical analyses were performed using appropriate parametric tests based on experimental design: two-tailed paired Student’s t-test for pairwise comparisons, one-way ANOVA for single-factor experiments, and two-way repeated measures ANOVA for time-course or multiple-factor studies. When ANOVA revealed significant main effects (p < 0.05), post hoc pairwise comparisons were conducted using Tukey’s honestly significant difference (HSD) test. All statistical computations were performed using SigmaPlot version 12.5 (Systat Software, San Jose, CA, USA).
Sample sizes were determined through a combination of preliminary pilot studies, established protocols from previous investigations,20–22,26 and statistical power analysis (α = 0.05, power = 0.90) using G*Power software (version 3.1.9.2). The threshold for statistical significance was maintained at p < 0.05 for all analyses.
Results
Peripheral nerve injury downregulates Oprm1 expression while upregulating MBD1, SUV39h1, and H3K9me3 in DRG, correlating with neuropathic pain development
To investigate the regulatory roles of DRG SUV39h1 and MBD1 in Oprm1 gene suppression following peripheral nerve injury, we examined the temporal expression patterns of MBD1 and SUV39h1 at both mRNA and protein levels in a well-established mouse model of neuropathic pain induced by unilateral L4 spinal nerve ligation (SNL 20 ). Quantitative RT-PCR analysis revealed a significant time-dependent reduction in Oprm1 mRNA levels in the ipsilateral L4 DRG at postoperative days 3, 7, and 14 compared to baseline (day 0), with no significant changes observed in sham-operated controls (Figure 1(a)). In contrast, mRNA expression of both Mbd1 and Suv39h1 exhibited progressive upregulation in the injured DRG, reaching peak levels at day 7 post-SNL (Figure 1(b) and (c)). Western blot analysis demonstrated corresponding increases in protein expression of MBD1, SUV39h1, and its catalytic product H3K9me3 in the ipsilateral L4 DRG following SNL injury (Figure 1(d)). These molecular changes were temporally associated with the development of neuropathic pain behaviors, as evidenced by: (1) mechanical allodynia, indicated by increased paw withdrawal frequency to von Frey filament stimulation (Figure 1(e)); (2) thermal hyperalgesia, reflected by decreased paw withdrawal latency to heat stimulation (Figure 1(f)); and (3) cold allodynia, demonstrated by reduced withdrawal latency to cold stimulation (Figure 1(g)). Motor function was consistent between mice that underwent SNL surgery and those that did not (Table S2). Notably, these behavioral changes were restricted to the ipsilateral hindpaw, with no significant alterations observed in contralateral paws of SNL-operated mice or either paw in sham-operated controls. These findings demonstrate that peripheral nerve injury induces a coordinated molecular response characterized by Oprm1 downregulation and MBD1/SUV39h1/H3K9me3 upregulation in the injured DRG, which correlates with the development of mechanical and thermal hypersensitivity in the affected limb.

Peripheral nerve injury downregulates Oprm1 expression while upregulating MBD1, SUV39h1, and H3K9me3 in DRG, correlating with neuropathic pain development: (a–c) Temporal changes in Oprm1, Mbd1, and Suv39h1 mRNA levels in the lesioned DRG following SNL surgery. L4 DRG tissues were collected from ipsilateral (Ips) and contralateral (Con) sides of sham and SNL mice at various time points before and after surgery. n = 6 mice per group, with three replicates. **p < 0.01 compared to the sham group at day 0, analyzed by one-way ANOVA with Tukey’s post hoc test. mRNA levels were normalized to Gapdh and quantified using RT-PCR. (d) Temporal changes in MBD1, SUV39h1, and H3K9me3 protein expression in the lesioned DRG after SNL surgery. L4 DRG tissues were collected from the ipsilateral side of SNL mice at baseline (Day 0) and 3, 7, and 14 days post-surgery. H3 served as a loading control. Left panel: Representative Western blot bands. Right panel: Statistical summary of densitometric analysis. n = 6 mice/group. **p < 0.01 compared to day 0, analyzed by one-way ANOVA with Tukey’s post hoc test. (e–g) Behavioral responses to mechanical (e), heat (f), and cold (g) stimuli on the ipsilateral and contralateral sides at different time points after SNL or sham surgery. n = 6 mice/group. **p < 0.01 compared to the contralateral side of the sham group (Sham-Con) in panels (e) and (f), or the ipsilateral side of the sham group (Sham-Ips) in panel (g) at the corresponding day 0 time point, analyzed by two-way ANOVA with Tukey’s post hoc test.
MBD1 knockout reverses SNL-induced epigenetic modifications and alleviates neuropathic pain
Peripheral nerve injury was found to upregulate MBD1, SUV39h1, and H3K9me3 expressions while downregulating MOR expression in the lesioned dorsal root ganglion (DRG). To investigate the involvement of these molecular changes in MOR suppression following nerve injury, we compared MBD1 knockout mice with wildtype controls at 7 days post-SNL lesion, analyzing gene and protein expressions in the DRG and assessing pain-related behaviors. Our results demonstrated that MBD1 knockout enhanced basal MOR expression at both transcriptional and translational levels, effectively preventing its downregulation in the ipsilateral L4 DRG post-SNL injury. While SUV39h1 expression remained unaffected by MBD1 knockout, both in basal conditions and following injury (Figure 2(a) and (b)), the knockout specifically blocked SNL-induced H3K9me3 upregulation in the lesioned DRG (Figure 2(b)). Behavioral analyses revealed that MBD1 knockout mice exhibited reduced sensitivity to mechanical stimuli (Figure 2(c), left panel) and increased thermal withdrawal thresholds at baseline (day 0; Figure 2(d), (e), left panel). Following SNL injury, these mice showed significant improvements in pain-related behaviors compared to wildtype controls, including reduced mechanical allodynia (Figure 2(c), right panel), attenuated heat hyperalgesia (Figure 2(d), right panel), and diminished cold allodynia (Figure 2(e), right panel) at day 7 post-injury. These findings indicate that MBD1 knockout specifically reverses SNL-induced H3K9me3 elevation and MOR suppression in injured DRGs, without affecting SUV39h1 expression patterns. Furthermore, the genetic ablation of MBD1 significantly attenuates neuropathic pain behaviors following peripheral nerve injury, highlighting its critical role in pain pathway regulation through epigenetic mechanisms.
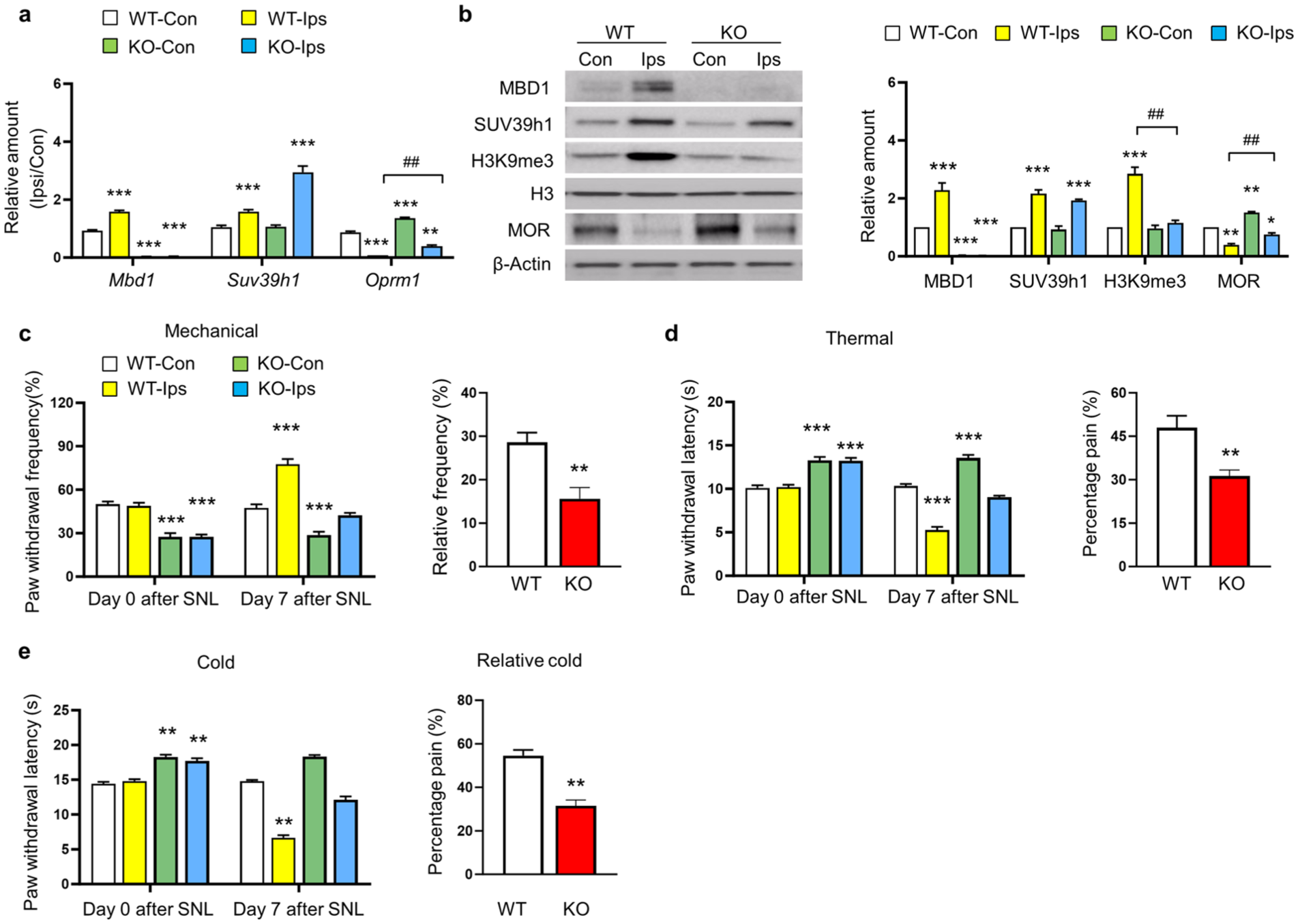
MBD1 knockout reverses SNL-induced epigenetic modifications and alleviates neuropathic pain. All experiments were conducted in wildtype (WT) or Mbd1-/- (KO) mice at 7 days post-SNL surgery. (a) mRNA expression of Mbd1, Suv39h1, and Oprm1 in ipsilateral (Ips) and contralateral (Con) L4 DRG of WT or Mbd1-/- mice at 7 days post-SNL injury. n = 4 replicates (8 mice per group). *p < 0.05, **p < 0.01, ***p < 0.001 compared to the WT-Con group; ##p < 0.01 compared to the WT-Ips group, analyzed by one-way ANOVA with Tukey’s post hoc test. (b) Protein levels of MBD1, SUV39h1, H3K9me3, and MOR in contralateral and ipsilateral L4 DRG of WT or Mbd1-/- mice at 7 days post-SNL surgery. Left: Representative Western blot bands. Right: Statistical summary of densitometric analysis. n = 8 mice/group (4 biological replicates). *p < 0.05, **p < 0.01 compared to the WT-Con group; ##p < 0.01 compared to the WT-Ips group, analyzed by one-way ANOVA with Tukey’s post hoc test. (c–e) Behavioral responses to mechanical (c), heat (d), and cold (e) stimuli before (day 0) and 7 days post-SNL surgery in contralateral and ipsilateral sides of WT or Mbd1-/- mice. Raw values (left) and relative frequency (right: value day 7 time-point – value baseline (day 0)) or percentage pain (right: value contralateral – value ipsilateral)/value contralateral *100 was shown. WT, wild type; KO, Mbd1-/- mice. n = 8 mice/group. The One-way ANOVA with post hoc Tukey test for Raw values and two-tailed unpaired Student’s t test for relative frequency or percentage pain, **p < 0.01 compared to the corresponding WT-Con group at the different time-points for raw values and **p < 0.01 compared to WT group for relative frequency or percentage pain.
Co-expression and interaction of MBD1 and SUV39h1 with Oprm1 mRNA in DRG Neurons
To elucidate the mechanisms underlying MBD1- and SUV39h1-mediated MOR suppression in lesioned DRG following nerve injury, we first examined the co-expression patterns of Mbd1, Suv39h1, and Oprm1 mRNA in DRG neurons of naïve mice. Using single-cell PCR analysis, as previously described, 20 we demonstrated that Mbd1, Suv39h1, and Oprm1 mRNA were co-expressed in individual DRG neurons across different size categories, including small, medium, and large neuronal cells (Figure 3(a)–(c)). To further investigate the potential molecular interaction between MBD1 and SUV39h1, we performed co-immunoprecipitation assays using naïve DRG tissues. Our results revealed that SUV39h1 antibody, but not IgG control, successfully pulled down MBD1 protein in wildtype mice, while no interaction was observed in Mbd1-/- mice (Figure 3(d)). This finding provides direct evidence for the physical interaction between MBD1 and SUV39h1 proteins at the DRG level under physiological conditions. These results collectively suggest that the MBD1-SUV39h1 protein complex may play a direct regulatory role in Oprm1 gene expression within DRG neurons. The co-expression of these molecular components in individual neurons and their physical interaction provide a potential mechanism for their coordinated regulation of MOR expression in the DRG.

Co-expression and Interaction of MBD1 and SUV39h1 with Oprm1 mRNA in DRG Neurons. (a–c) Oprm1 mRNA co-expression with Mbd1 and Suv39h1 mRNA in individual large (a), medium (b), and small (c) neurons from the naïve mice DRGs, respectively. n = 3 repeats for single cell PCR. (d) The binding of SUV39h1 to MBD1 in the DRG neuron. MBD1 was immunoprecipitated by rabbit anti-SUV39h1 antibody in the nuclear fraction of DRG neurons from the wildtype mice, but not Mbd1-/- mice. n = 3 repeats for Co-immunoprecipitation.
MBD1-dependent recruitment of SUV39h1-mediated H3K9me3 formation suppresses Oprm1 gene transcription in DRG neurons
To elucidate the molecular mechanisms underlying MBD1- and SUV39h1-mediated regulation of MOR expression in DRG neurons, we performed chromatin immunoprecipitation (ChIP) assays targeting five distinct fragments of the Oprm1 gene promoter. Our analysis revealed that both SUV39h1 and H3K9me3 specifically bound to the −340 to −200 bp region of the Oprm1 promoter in naïve mouse DRG, with no detectable binding at other promoter fragments (Figure 4(a) and (b)). This finding aligns with our previous report demonstrating MBD1 binding to the same promoter region. 20 Following SNL injury, we observed a 3.0-fold increase in SUV39h1 binding activity at this promoter region in ipsilateral DRG of wildtype mice, while MBD1-deficient mice showed a 0.46-fold reduction compared to contralateral DRG of wildtype controls at day 7 post-injury (Figure 4(c)). MBD1 knockout effectively reversed the injury-induced increase in SUV39h1 binding activity. Similar binding patterns were observed for H3K9me3 at the Oprm1 promoter (Figure 4(d)), suggesting that the recruitment of SUV39h1 and H3K9me3 to this regulatory region is partially dependent on MBD1 expression. To further investigate the functional relationship between MBD1 and SUV39h1, we manipulated their expression in cultured DRG neurons using AAV5-mediated MBD1 overexpression (AAV5-Mbd1) and SUV39h1-specific siRNA (siSuv39h1). AAV5-Mbd1 transduction significantly reduced Oprm1 mRNA levels compared to AAV5-GFP controls (Figure 4(e)), while siSuv39h1 transfection upregulated Oprm1 expression relative to non-targeting siRNA controls (Figure 4(f)). Co-transduction experiments revealed that the effects of MBD1 overexpression were abolished in the presence of SUV39h1 knockdown (Figure 4(e)). Consistent with these findings, SUV39h1 knockdown decreased H3K9me3 levels while increasing MOR protein expression in cultured DRG neurons (Figure 4(f)). These results demonstrate that MBD1 and SUV39h1 collaboratively suppress MOR expression in lesioned DRG through increased H3K9me3 enrichment at specific Oprm1 promoter regions, leading to chromatin condensation and transcriptional silencing. This epigenetic regulatory mechanism provides a molecular basis for MOR downregulation following peripheral nerve injury.

MBD1-dependent recruitment of SUV39h1-mediated H3K9me3 formation suppresses Oprm1 gene transcription in DRG neurons. (a) ChIP assays using SUV39h1-specific antibody (not IgG control) demonstrated selective enrichment of the R1 fragment (−340/−200 bp) within the 5′-UTR and promoter region of the Oprm1 gene in murine DRGs, while other regions (R2: −268/−138 bp; R3: −63/+133 bp; R4: +163/+357 bp) showed no significant binding. Similar ChIP analysis using H3K9me3-specific antibody confirmed enrichment of the R1 fragment in the Oprm1 promoter region. M: DNA ladder marker; Input: total chromatin fragments; R: genomic region. Experiments were performed in triplicate. (b) Quantitative analysis of SUV39h1 binding to the R1 fragment in L4 DRGs from wildtype and Mbd1-/- mice 7 days post-SNL surgery. Contralateral and ipsilateral DRGs were analyzed separately (n = 4 replicates, 8 mice/group). Statistical significance was determined by one-way ANOVA with Tukey’s post-hoc test (*p < 0.01 vs WT-Con; ##p < 0.01 vs WT-Ips). (c) H3K9me3 enrichment at the −340/−200 bp region of the Oprm1 promoter in L4 DRGs under identical experimental conditions (**p < 0.01 vs WT-Con; ##p < 0.01 vs WT-Ips). (d) Quantitative RT-PCR analysis of Mbd1, Suv39h1, and Oprm1 mRNA levels in cultured DRG neurons following transduction with AAV5 constructs and siRNA treatments: AAV5-Gfp + siNC, AAV5-Mbd1 + siNC, AAV5-Gfp + siSuv39h1, or AAV5-Mbd1 + siSuv39h1 (n = 3 replicates, 6 mice/group). Statistical analysis by one-way ANOVA with Tukey’s test (**p < 0.01 vs AAV5-Gfp + siNC; ##p < 0.01 vs AAV5-Mbd1 + siNC). (e) Western blot analysis of SUV39h1, H3K9me3, and MOR protein expression in cultured DRG neurons transfected with siNC or siSuv39h1. Representative immunoblots (left panel) and quantitative densitometric analysis (right panel) are shown (n = 3 replicates, 6 mice/group). Statistical significance was determined by two-tailed paired t-test (**p < 0.01 vs siNC).
Discussion
Although the roles of DNA methylation and histone modification in gene expression regulation under neuropathic pain conditions have been extensively studied over the past decades, the mechanisms by which DNA methylation and histone modification cooperatively regulate pain-associated factors in the context of peripheral nerve injury remain poorly understood. This study provides evidence that SUV39h1-mediated H3K9me3 formation plays a critical role in the epigenetic downregulation of the Oprm1 gene in the DRG following peripheral nerve injury, a process that requires MBD1 recruitment. Notably, genetic ablation of MBD1 reversed the downregulation of MOR, attenuated the increase in H3K9me3 levels, and alleviated pain hypersensitivity following nerve injury, despite the elevated expression of SUV39h1. Mechanistically, peripheral nerve injury enhanced the binding of SUV39h1 and H3K9me3 to the Oprm1 gene promoter, effects that were abolished in the absence of MBD1. These findings suggest that MBD1 recruits SUV39h1 to specific sites on the Oprm1 gene promoter, where it catalyzes H3K9me3 formation, leading to chromatin condensation and subsequent transcriptional silencing of MOR expression.
SUV39h1, a critical regulator of gene expression, plays a significant role in the generation and progression of nerve lesion-induced neuropathic pain. Our previous study demonstrated that SUV39h1 contributes to nerve lesion-induced pain hypersensitivity, potentially through the regulation of MOR expression in the injured DRG. 13 However, the precise mechanisms underlying this process remain unclear. In this study, we revealed that SUV39h1/H3K9me3 specifically binds to a single fragment (−340/−200 bp) within the Oprm1 gene promoter, out of four examined fragments (−450 bp to +357 bp). This finding aligns with our earlier data showing that MBD1 also specifically binds to the same region (−340/−200 bp) of the Oprm1 promoter. 20 Notably, genetic ablation of MBD1 reduced the binding activity of SUV39h1 and H3K9me3 to this region and reversed the spinal nerve ligation (SNL)-induced increase in their binding to the Oprm1 promoter. These results indicate that the binding of SUV39h1 and H3K9me3 to this specific promoter region is dependent on MBD1. Collectively, these findings suggest that the increased enrichment of histone H3 lysine trimethylation (H3K9me3), mediated by SUV39h1, is localized to a specific site within the 5′-untranslated regions of the Oprm1 promoter following SNL. Importantly, MBD1, SUV39h1, and the resulting H3K9me3 modification share the same binding site on the Oprm1 promoter in the injured DRG under neuropathic pain conditions.
MOR is predominantly expressed in DRG neurons, particularly in calcitonin gene-related peptide (CGRP)-positive peptidergic small neurons and pain fibers.27,28 Activation of MOR by endogenous opioids inhibits neurotransmitter release from these fibers and reduces neuronal excitability.29,30 Downregulation of MOR in the DRG enhances neurotransmitter release from primary afferents,31,32 and diminishes the analgesic efficacy of opioids in neuropathic pain patients. 18 The observed decrease in Oprm1 gene expression in the injured DRG following peripheral nerve injury likely involves multiple epigenetic mechanisms. In addition to SUV39h1-mediated H3K9me3, other epigenetic processes, such as DNA methylation and demethylation, may also contribute to the downregulation of MOR expression in the injured DRG after peripheral nerve trauma.26,33 Previous studies have shown that SUV39h1 interacts with other DNA methylation regulators to coordinate DNA and histone methylation.34,35 This study further demonstrates that the nerve lesion-induced upregulation of SUV39h1, a histone lysine methyltransferase, interacts with MBD1, which is essential for the transcriptional silencing of the Oprm1 gene in the injured DRG following SNL.
Our previous studies have shown that MOR co-localizes with SUV39h1 and MBD1 in DRG neurons.13,20 MBD1 is primarily expressed in CGRP-positive small peptidergic neurons of the DRG. Importantly, MBD1-deficient mice do not exhibit developmental defects, loss of sensory neurons, or abnormal morphological changes in the DRG or spinal cord dorsal horn, including neuronal and satellite cells, compared to wildtype mice. 20 Therefore, in this study, we focused on the molecular and behavioral changes in the ipsilateral and contralateral sides of wildtype and Mbd1-/- mice following SNL surgery, as no significant differences were observed between the ipsilateral and contralateral sides of sham-operated mice.
In conclusion, this study provides evidence that SUV39h1-mediated H3K9me3 contributes to the transcriptional repression of the Oprm1 gene in the injured DRG following peripheral nerve trauma, a process that requires MBD1 recruitment. Notably, MBD1 knockout not only reversed the downregulation of MOR mRNA and protein in the DRG but also attenuated the nerve lesion-induced upregulation of SUV39h1 and the associated enrichment of H3K9me3 at the Oprm1 promoter. Furthermore, MBD1 deficiency alleviated nerve lesion-induced pain hypersensitivity.
Our findings align with accumulating evidence that neuroimmune interactions and epigenetic regulation play central roles in neuropathic pain development.36,37 Neuroinflammation triggered by nerve injury – characterized by immune cell activation and pro-inflammatory cytokine release – is recognized as a critical upstream event driving epigenetic reprogramming of pain-related genes. 37 Epigenetic modifications, including histone changes such as H3K9me3, have been extensively observed in both peripheral and central nervous systems during chronic pain and regulate the expression of multiple pain-related genes, including MOR.38,39 The downregulation and functional loss of spinal MOR gene expression following nerve injury constitute major factors limiting morphine efficacy. 39 The MBD1-SUV39h1-H3K9me3 pathway identified here offers a novel molecular mechanism explaining how neuroinflammation epigenetically mediates the downregulation of key genes such as Oprm1, thereby sustaining pain.
Considering that cytokines released during neuroinflammation (e.g. MCP-3 and IL-6) regulate various epigenetic enzymes and reader proteins,36,37 a crucial next step is to determine whether these aberrant cytokines serve as upstream signals activating or modulating the MBD1-SUV39H1 axis within the dorsal root ganglion, as newly identified in our study. Therefore, future research will focus on elucidating how these cytokines influence the expression, activity, and promoter enrichment of axis components at the Oprm1 locus, aiming to clarify the upstream mechanisms governing this epigenetic silencing cascade.
These findings suggest that MBD1 and SUV39h1 represent promising therapeutic targets for the prevention and treatment of neuropathic pain.
Supplemental Material
sj-docx-1-mpx-10.1177_17448069251389416 – Supplemental material for MBD1-dependent recruitment of SUV39h1 induces H3K9me3-mediated transcriptional silencing of Oprm1 in dorsal root ganglia after peripheral nerve injury
Supplemental material, sj-docx-1-mpx-10.1177_17448069251389416 for MBD1-dependent recruitment of SUV39h1 induces H3K9me3-mediated transcriptional silencing of Oprm1 in dorsal root ganglia after peripheral nerve injury by Hengwei Sheng, Hualei Gong, Xiaolin Liu, Wenbo Wu, Helena Soares da Silva, Sen Huang and Kai Mo in Molecular Pain
Footnotes
Acknowledgements
The authors express their gratitude to Dr. Yuan-Xiang Tao and Dr. Shaogen Wu (Rutgers State University of New Jersey) for generously providing Mbd1-/- mice and full-length Mbd1 cDNA plasmids. We also acknowledge the valuable contributions of recent studies that have advanced our understanding of epigenetic mechanisms in neuropathic pain, which have informed the interpretation of our findings.
Author contributions
Hengwei Sheng: Investigation, Formal analysis, Visualization, Methodology, Writing – Review & Editing. Hualei Gong: Investigation, Methodology, Formal analysis, Data curation, Writing – Review & Editing. Xiaolin Liu: Investigation, Writing – Review & Editing. Wenbo Wu: Investigation, Writing – Review & Editing. Helena Soares da Silva: Investigation, Writing – Review & Editing. Sen Huang: Methodology, Investigation, Writing – Review & Editing. Kai Mo: Conceptualization, Writing – Original Draft, Writing – Review & Editing, Formal analysis, Supervision, Project administration, Funding acquisition.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the National Natural Science Foundation of China (Grant No. 82471228) awarded to Kai Mo, Guangdong Basic and Applied Basic Research Foundation (Grants No. 2021A1515220006 and 2022A1515010410) to Kai Mo, and Medical Scientific Research Foundation of Guangdong Province of China (Grant No. 2020111195224883) to Hengwei Sheng.
Resource availability
The data supporting this article are included as part of the supplementary information. Details regarding the sources and methods for data obtained from public resources are provided in the Materials & methods section. Requests for further information should be directed to and will be fulfilled by the lead contact, Kai Mo (
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.