
Abstract
G protein-coupled kinase (GRK) 6 is a member of the GRK family that mediates agonist-induced desensitization and signaling of G protein-coupled receptors (GPCRs), thus involving in a wide variety of processes including inflammation and nociception. Recent studies have indicated that chemokines play an important role in chronic pain via increased expression of respective GPCRs. This study was designed to investigate the role of GRK6 and its interaction with substrate chemokine receptors in dorsal root ganglion (DRG) in a rat model of neuropathic pain induced by chronic constriction injury (CCI). Following induction of CCI, GRK6 expression was significantly downregulated in rat DRGs at L4-L6 segments. Overexpression of GRK6 using lentiviral-mediated production strategy via sciatic nerve injection markedly attenuated mechanical allodynia and thermal hyperalgesia in CCI rats. Overexpression of GRK6 also drastically reversed the hyperexcitability of DRG neurons innervating the hind paw and suppressed the enhanced expression of CXCR2 in DRGs of CCI rats. In addition, co-immunoprecipitation, immunofluorescence, and correlation analysis supported the interaction between GRK6 and CXCR2. These results suggest that GRK6 might be a key molecular involved in peripheral mechanism of neuropathic pain and that overexpression of GRK6 might be a potential strategy for treatment for neuropathic pain through inhibition of CXCR2 signal pathway.
Introduction
*Yuan Zhou and Rong-Ji Li contributed equally to this work.
Merging evidence suggests that abundant mediators are involved in neuropathic pain function via activating G protein-coupled receptors (GPCRs) expressed in sensory neurons. 6 G protein-coupled kinases (GRKs) regulate cellular signaling by promoting desensitization of agonist-occupied GPCRs. GRKs-mediated phosphorylation of the activated GPCR promotes binding of arrestin proteins and rapid uncoupling from the G proteins, leading to GPCR desensitization and internalization. 7 Increasing evidence has shown that intracellular levels of GRK determine GPCR sensitivity. 8 GRKs also interact directly with a variety of downstream signaling molecules, thereby regulating cellular signaling pathways.9,10 GRK6, as a ubiquitously expressed member of the GRK family, has been shown to play a critical role in inflammatory diseases.11,12 It has been reported that desensitization of many GPCRs, such as CGRP receptors, the BLT1 receptors for the leukotriene LTB4, and the chemokine receptors, are all regulated by GRK6.13–15 Several reports have demonstrated the involvement of GRK6 in pain modulation.16–18 More recently, we have reported that chronic neuropathic pain is associated with decreased levels of the GRK6 in spinal neurons. 19 But it is largely unknown whether the peripheral GRK6 in dorsal root ganglion (DRG) and which specific substrate GPCR signaling pathway play a role in neuropathic pain.
Chemokines, a family of small secreted cytokines, have been demonstrated to regulate chronic pain via neuroinflammation at different anatomical locations, including nerve, DRGs, spinal cord, and the brain. 20 They are classified in different families according to the number and localization of amino terminal cysteine residues. 21 Of high interest, some members of the CXC chemokine family, with a glutamate–leucine–arginine (ELR+) domain in the amino terminal portion of the molecule, are known notably for their ability to attract neutrophils to the site of inflammation. 22 The representative components of this subfamily, CXCL1 (keratinocyte-derived chemokine, KC; GROa), CXCL8 (interleukin-8, IL-8), and CXCL12, have been revealed an upregulation in several animal models following nerve injury and implicated in the development and maintenance of neuropathic pain.23–25 The biological effects of chemokines are mediated via G protein-coupled chemokine receptors, including CXCR1, CXCR2, and CXCR4. Compelling evidence demonstrated that expression of these chemokine receptors is significantly upregulated during both early-phase and late-phase of chronic pain and that blocking these receptors can reverse pain-related behaviors.26–28 However, the possible molecular mechanism underlying the upregulation of chemokine receptors-mediated nociceptive sensation remains to be elucidated.
Previous studies have reported that GRK6 may phosphorylate distinct chemokine receptors in mediating different cellular functions.29,30 In this study, we sought to explore roles of GRK6 in DRGs and to determine the specific chemokine receptor signaling pathway mediated by GRK6 under neuropathic pain conditions. To that end, we investigated the temporal expression and cellular location of GRK6 in the DRGs in a well-established CCI model. We then tested the pain behaviors and measured the excitability of hind paw innervating DRG neurons after injection of GRK6 over-expression lentivirus. We showed GRK6 was downregulated following CCI and that overexpression of GRK6 attenuated the pain responses and suppressed the upregulation of CXCR2. Collectively, our findings represented a peripheral role of GRK6 in CCI model and supported a new mechanism underlying the activation of CXCR2 signal pathway in neuropathic pain.
Methods and materials
Induction of CCI-induced pain model
Experiments were performed on adult Sprague-Dawley (SD) rats (200 ± 20 g, male). Animals were housed under controlled conditions (24℃ ± 2℃, 12:12-h alternating light–dark cycle) with free access to food and water. All experiments were approved by the Institutional Animal Care and Use Committee of Soochow University and were in accordance with the guidelines of the International Association for the Study of Pain. The surgical procedure was carried out aseptically under anesthetized with chloral hydrate (10% solution, ip). CCI was performed at the mid-thigh level of the right hind leg by loosely ligating the common sciatic nerve as described previously. 31 In brief, proximal to the sciatic trifurcation, four ligatures (4.0 chromic gut) were loosely tied around the isolated sciatic nerve with a 1.0 to 1.5 mm interval between each of them. The sciatic nerves of Sham-operated rats were identically exposed and manipulated but no ligation was made. Skin wound was sutured with 7.0 nylon suture.
Pain behavior measurements
Male rats (CCI group, n = 8; Sham group, n = 8) were used for assessment of mechanical allodynia and thermal hyperalgesia of the hind paws 3 days before surgery and 3, 7, 14, 21 days after surgery by an investigator in a blinded manner. Mechanical allodynia was measured as changes in hind paw mechanical withdrawal thresholds (PWT) by using von Frey filaments (VFF) as previously described. 32 Briefly, rats were placed in individual plastic boxes and allowed to acclimate for at least a half hour. A series of calibrated VFFs (0.55, 0.93, 1.61, 1.98, 2.74, 4.87, 7.37, 11.42, 15.76, and 20.30 g) were applied perpendicularly to the plantar surface of the hind paw with sufficient force to bend filaments for 1 to 2 s. A next greater force VFF was used in the presence of a response, and a next lower force VFF was employed in the absence of a response. The cut-off strength of VFF was set at 20.30 g to avoid unnecessary tissue damage during the tests. The modification of the “up-down” calculating method was applied to determine the value at which paw withdrawal occurred 50% of the time. Each trial was repeated three times at approximately 5-min intervals, and the average value was PWT.
Thermal hyperalgesia of the hind paw was measured by a well-established method previously. 33 In brief, rats were placed on the clear glass surface of acrylic chambers and allowed to acclimatize for a half hour. A radiant heat source focused onto the plantar surface of the hind paw. The intensity of the thermal stimulus was adjusted to derive an approximately 10.0 s average baseline of paw withdraw latency (PWL) in normal animals. Measurements of PWL were determined by a timer that was started by activation of a heat source and stopped when the hind paw withdrawal was detected with a photodetector. A maximal cut-off time of 20 s was applied to prevent tissue damage during the heat tests. The mean PWL was obtained by averaging the latency of three measurements with a 10-min interval between consecutive tests.
Western blotting
Rats were sacrificed by an overdose of chloral hydrate. Lumbar DRGs (L4-L6) from the ipsilateral side were quickly dissected out and frozen in liquid nitrogen. Each sample was lysed and centrifuged at 12,000 r/min for 30 min at 4℃ to collect the supernatant; 20 mg of protein was fractionated on 10% polyacrylamide gels (Bio-Rad, Hercules, CA, USA) and then transferred to polyvinyl difluoride (PVDF) membranes (Bio-Rad, USA) for 2 h at 200 mA. The membranes were blocked in TBS (50 mM Tris-Base, 133 mM NaCl, pH = 7.4) containing a 5% dilution of non-fat milk powder for 2 h at room temperature (RT) and then incubated with primary antibody (anti-GRK6 at 1:500, Santa Cruz Biotechnology, USA; CXCR1 at 1:500, Santa Cruz Biotechnology, USA; anti-CXCR2 at 1:100, Boster, P.R. China; anti-CXCR4, Santa Cruz Biotechnology, USA; anti-GAPDH at 1:1000, Goodhere, China; anti-β-actin at 1:1000, Multi-Sciences, P.R. China) in TBS containing 1% milk at 4℃ overnight. After washing in TBST (0.5% Tween-20 in TBS), the PVDF membranes were incubated with HRP-conjugated secondary antibodies (1:4000, Multi Sciences Biotech Co., P.R. China) in TBS and 1% milk for 2 h at RT. Bands were visualized using ECL (Biological Industries, P.R. China) and exposed to Kodak X-ray films. Films were scanned and band intensities of target proteins were analyzed using Optic Quant software (ImageJ, NIH, USA).
Immunofluorescence study
One week after surgical procedure and Dil or lentiviral injection, the CCI or sham rats were perfused transcardially with 300 mL 0.9% saline followed by iced 4% paraformaldehyde. L4-L6 DRGs were removed and postfixed for 3 h and then immersed in the increasing phosphate-buffered sucrose solution for cryoprotection: 20% overnight and 30% for three days at 4℃. For double labeling, 10-mm cryosections of DRG were incubated with polyclonal primary antibody for GRK6 (anti-rabbit, 1:200, Santa Cruz Biotechnology, USA), CXCR1 (anti-rabbit, 1:100, Santa Cruz Biotechnology, USA), CXCR2 (anti-rabbit,1:50, Boster, P.R. China), CXCR4 (anti-rabbit, 1:100, Santa Cruz Biotechnology, USA), and specific marker proteins such as a rabbit polyclonal CGRP antibody (1:100, Chemicon), FITC-conjugated Bandeiraea simplicifolia BS-I isolectin B4 (IB4, 5 µg/ml, Sigma), mouse monoclonal anti-GFAP (1:200, Sigma), β-tubulin (1:200, Millipore) overnight at 4℃. On the following day, a mixture of secondary antibody with Alexa Fluor 488 and 355 were added and incubated for 2 to 3 h in a dark room at RT. Negative control (NC) was performed by omitting primary antibody. Sections were viewed with filter cubes appropriate for Alexa 355, 488, and DiI (rhodamine filter). The stained sections were viewed with a Leica fluorescence microscope (Germany). Images were captured and analyzed by Metaview software as described previously. 34
Cell retrograde labeling
DRG neurons innervating the hind paw were labeled by retrograde tracing using 1,1
Dissociation of DRG neurons and patch-clamp recordings
Changes in neuronal excitability were determined using whole-cell patch-clamp recording techniques as described previously. 34 In brief, the ipsilateral L4-L6 DRGs were dissected out one week after Dil injection. Three DRGs were then transferred to an ice-old and oxygenated fresh dissecting solution containing collagenase D (1.8 to 2.0 mg/mL; Roche, Indianapolis, IN, USA) and trypsin (1.2 to 1.5 mg/mL; Sigma, St Louis, MO, USA) and incubated for 1.5 h at 34.5℃. The dissecting solution contains (in mmol/L) 130 NaCl, 5 KCl, 2 KH2PO4, 1.5 CaCl2, 6 MgSO4, 10 glucose, and 10 HEPES, pH = 7.2; osmolarity = 305 mOsm. The ganglia were then transferred to 2 mL of the dissecting solution with DNase (0.5 mg/mL, Sigma, USA). A single cell suspension was subsequently obtained by repeated trituration through flame-polished glass pipettes. Cells were plated onto acid-cleaned glass coverslips. DiI-labeled neurons were identified by their fluorescence under a fluorescence microscope (Olympus IX71, Japan). Normal external solution contains (in mmol/L) 130 NaCl, 5 KCl, 2 KH2PO4, 2.5 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, with pH adjusted to 7.2 with NaOH; 295 to 300 mOsm. Patch-clamp pipettes had a resistance of 4 to 7 MΩ when filled with the pipette solution, which contains (in mmol/L) 140 potassium gluconate, 10 NaCl, 10 HEPES, 10 glucose, 5 EGTA, and 1 CaCl2, pH = 7.25 adjusted with KOH; 292 mOsm. The resting membrane potential (RP) and action potentials (APs) were recorded under current-clamp configuration using an EPC10 patch-clamp amplifier (HEKA, Germany). Neurons with a RP more depolarized than −40 mV were excluded in the data analysis.
Drug application
SB225002 (purchased from Selleck, USA), a selective inhibitor of CXCR2, was freshly prepared in DMSO (Sigma). Seven days after CCI application, SB225002 was administered intrathecally at different doses (20, 40, and 60 µg in 10 µL DMSO) for the behavioral measurements. A guiding needle (18 G) was passed between the lumbar vertebrae 5 and 6 to enter the intrathecal space and the SB225002 solution was slowly injected.
Lentiviral vectors production and infection
The full-length of GRK6 was cloned from rat brain cDNA library and sub-cloned into pENTRTM/D-TOPO vector. After recombined the enter clone pENTRTM/D-TOPO GRK6 with the lentiviral destination vector (LR and BP) by the recombined enzymes, the recombined lentiviral destination vector and packaging plasmids (Shanghai Gene Chem Co. Ltd., P.R. China) were con-transfected 293FT cells to generate virus stocks of Lenti-GRK6 (LV-GRK6) or NC lentivirus (LV-NC) after 72 h. Estimated titers of the concentrated LV-NC/LV-GRK6 were 1.25 × 108 transducing units per milliliter (TU/mL). Adult SD rats weighing 180 to 220 g were used. The animals were anesthetized by isoflurane (1-chloro-2, 3, 4-trifluoroethyl ether, AErrane) with oxygen. Operations were performed using an operating microscope under aseptic conditions. The sciatic nerves were exposed in the mid-gluteal region through a gluteal muscle-splitting incision. A 27-G needle was inserted longitudinally into the nerve and 4 µL volume of LV-NC/LV-GRK6 was injected slowly using a micromanipulator. The nerves were anatomically repositioned, and the skin was closed with 7.0 nylon sutures.
Co-immunoprecipitation (Co-IP)
Each sample was lysed and centrifuged at 12,000 r/min for 30 min at 4℃ to collect the supernatant. The concentration of protein in the supernatant was determined by the Bradford method using BSA as standard (BioRad); 500 µg extract was incubated with either 5 µg anti-GRK6 antibody (anti-mouse, 1:200, Santa Cruz Biotechnology, USA) or pre-immune IgG in 500 μl of Co-IP buffer (Active motif) at 4℃ overnight with rotation. The immunoprecipitates were isolated on Protein A beads and separated by 10% SDSPAGE followed by transferring to PVDF membrane. The membrane was probed with antibodies against GRK6 and CXCR2 (anti-rabbit, 1:100, Boster, P.R. China).
Quantification and statistics
All data are expressed as means ± SEM. Statistical testing was carried out using a stepwise procedure depending upon the number of groups being compared. Normality was first examined for all data before analysis. When only two means were involved in a comparison, a two-tailed t test with unequal variances was used. When more than two means were involved, a one-way analysis of variance (ANOVA) or Friedman ANOVA was first carried out to obtain a global test of the null hypothesis. If the global p value for the test of the null hypothesis was <.05, post hoc comparisons between the different groups using Mann–Whitney test or Dunn’s post hoc test were performed. A comparison was considered statistically significant when a p value was <.05.
Results
CCI induces persistent neuropathic pain and downregulates GRK6 expression in DRGs
CCI significantly produced mechanical allodynia and thermal sensitivity of the injured hind paw compared with the Sham-operated rats. In CCI group, a persistent decreased mechanical paw withdrawal threshold (PWT) on the ipsilateral side was observed from 3 days through 21 days after surgery, and the maximal mechanical allodynia was shown at seven days (Figure 1(a), n = 8 for each group, *p < .05 vs. Sham, Mann–Whitney test following Friedman ANOVA). Similarly, withdrawal latencies of the affected hind paw to radiant heat were significantly reduced from post-operative day 3 to day 21 compared with those of the Sham group (Figure 1(b), n = 8 for each group, *p < .05 vs. Sham, Mann–Whitney test following Friedman ANOVA). To investigate the temporal expression of GRK6 in L4-L6 DRGs following CCI, DRGs from ipsilateral side at 3, 7, 21 days were analyzed by Western blot assay. GRK6 protein expression was significant reduced from day 3 to day 21 after surgery, and the lowest level appeared at day 7 (Figure 1(c), n = 3 for each group, *p < .05 vs. Sham). In addition, we determined the cellular location of GRK6 expression in the DRGs using immunofluorescence staining. Double staining showed that GRK6-like immunoreactivities were mostly in neurons marked with β-tubulin and not expressed in glial cells labeled with GFAP. Co-location of GRK6 with IB4 and CGRP demonstrated that GRK6 expressed in both non-peptide and peptide containing DRG neurons with unmyelinated fibers (Figure 1(d)).
Enhanced neuropathic pain and decreased expression of GRK6 in CCI rats. (a) CCI induces a significant decrease in PWT from 3 days to 21 days after surgery in the ipsilateral side. *p < .05, compared with Sham group. (b) The development of heat hyperalgesia from 3 days to 21 days was induced by CCI in the ipsilateral side. *p < .05, compared with Sham group. (c) Expression of GRK6 protein in DRGs was decreased from 3 days to 21 days after surgery in the ipsilateral side with the lowest level seven days after CCI. *p < .05, compared with Sham group. (d) GRK6 mainly expressed in neurons co-localized with β-tubulin, IB4, and CGRP in DRGs, but not in glial cells stained with GFAP. Bar = 75 µm.
Overexpression of GRK6 attenuates the mechanical allodynia and heat hyperalgesia
To further explore the role of GRK6 in the peripheral regulation of neuropathic pain induced by CCI, we next determined whether administration of GRK6-LV suppressed the mechanical hyperalgesia and heat hyperalgesia. Figure 2(a) showed that transgene expression could be achieved in the corresponding DRG after injection of lentiviral carrying the green fluorescent protein (LV-GFP) into the sciatic nerves (Figure 2(a), right). The GRK6-LV efficiency in DRG was examined by Western blot assay (Figure 2(b)). Before LV administration, PWT and PWL in three groups (CCI, NC-LV, and GRK6-LV) had no significant difference (Figure 2(c) and (d), PRE). However, GRK6 overexpression by lentiviral injection on sciatic nerve greatly enhanced PWT (Figure 2(c), *p < .05 vs. NC-LV, Mann–Whitney test following Friedman ANOVA) and PWL (Figure 2(d), *p < .05 vs. NC-LV, Mann–Whitney test following Friedman ANOVA) in rats with CCI.
Suppression of neuropathic pain by GRK6 overexpression. (a) GFP image showed the successfully infection of lentiviral in DRG by sciatic nerve injection. (b) Western blot analysis presented the enhanced expression of GRK6 in DRG from CCI rats after GRK6 lentiviral injection. (c) GRK6 lentiviral injection, rather than NC lentiviral, markedly increased PWT on the CCI rats. *p < .05, compared with CCI group. (d) GRK6 lentiviral injection, rather than NC lentiviral, markedly increased PWL on the CCI rats. *p < .05, compared with CCI group. LV-GFP: lentiviral carrying the green fluorescent protein; CCI: chronic constriction injury; NC-LV: Negative control lentivirus; PWL: paw withdraw latency; PWT: paw mechanical withdrawal thresholds.
CCI enhances the excitability of DRG neurons
To determine whether primary sensory afferents are involved in the development of hypersensitivity in CCI rats, we measured the excitability of hind paw innervating DRG neurons from Sham and CCI rats. Hind paw innervating DRG neurons were labeled by the fluorescent dye DiI injected into the right hind paw at five different points (Figure 3(a)). The small- and medium-sized DRG neurons were used in this study because they are the primary sensory neurons responsible for pain sensation.
35
Under current-clamp conditions, a total of 22 cells were recorded. RPs were −48.8 ± 1.0 mV (n = 11) and −42.2 ± 0.5 mV (n = 11) for hind paw innervating DRG neurons isolated from Sham and CCI rats, respectively. Therefore, CCI significantly depolarized RP (Figure 3(b); *p < .05 compared with Sham, Mann–Whitney test). The AP thresholds were −26.9 ± 1.0 mV (n = 11) and −31.0 ± 1.7 mV (n = 11) for Sham and CCI rats, respectively. CCI remarkably hyperpolarized the AP threshold (Figure 3(c); *p < .01 compared with Sham, Mann–Whitney test). The rheobase measurements were 77.3 ± 10.5 (n = 11) and 25.9 ± 6.0 pA (n = 11) for Sham and CCI rats, respectively. CCI also markedly reduced rheobase (Figure 3(d); *p < .05, compared with Sham, two-sample t test). In addition, CCI resulted in a dramatic increase in the numbers of APs evoked by 100 and 300 pA ramp current stimulation (Figure 3(e) to (h); *p < .05 compared with Sham, two-sample t test). The numbers of APs evoked by 100 pA ramp current stimulation were 2.8 ± 1.0 (n = 11) and 9.6 ± 1.3 (n = 11) for Sham and CCI rats, respectively. The numbers of AP evoked by 300 pA ramp current stimulation were 8.5 ± 1.1 (n = 11) and 15.0 ± 2.0 (n = 11) for Sham and CCI rats, respectively. CCI significantly enhanced the numbers of APs in response to ramp current stimulation.
Enhanced excitability of DRG neurons in CCI rats. (a) Bright-field (right) and DiI fluorescence (left) images of acutely dissociated DRG neurons. A hind paw innervating DRG neuron is shown in red (arrow). Bar = 50 µm. (b) CCI significantly depolarized the resting membrane potential (RP) of hind paw innervating DRG neurons. *p < .05 vs. Sham. (c) CCI notably hyperpolarized the action potential (AP) threshold. *p < 0.05 vs. Sham. (d) CCI also markedly decreased rheobase. *p < .05 vs. Sham. (e, g) Examples of APs by 100 and 300 pA ramp current injection from Sham and CCI rats. (f, h) Bar graph showed a significant increase in numbers of APs evoked by 100 and 300 pA ramp current stimulation in CCI rats. *p < .05 vs. Sham. RP: resting membrane potential; AP: action potential; CCI: chronic constriction injury.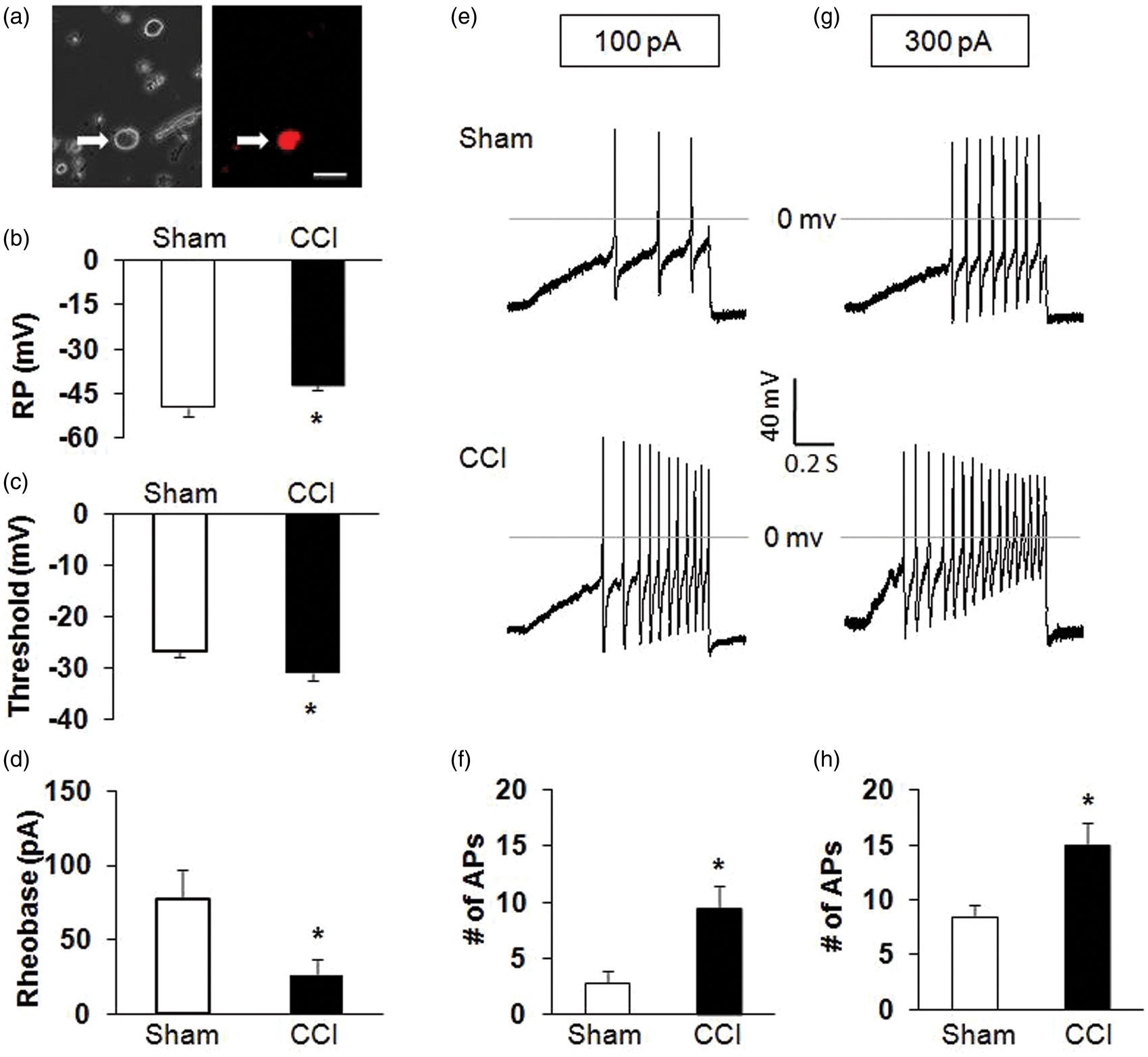
Overexpression of GRK6 reverses CCI-induced hyperexcitability of DRG neurons
Because GRK6 overexpression attenuates the mechanical allodynia and heat hyperalgesia in CCI rats, we next investigated whether GRK6 overexpression affects the excitability of hind paw innervating DRG neurons of CCI rats. Under current-clamp conditions, a total of 12 cells were recorded from three CCI rats treated with GRK6-LV injection and a total of 12 cells were from three CCI rats treated with NC-LV. Only GRK6-LV-infected (green, stained with GFP) hind paw innervating DRG neurons (red, Dil fluorescence) were measured in the present study (Figure 4(A)). RPs were −43.9 ± 1.0 mV (n = 12) and −48.2 ± 0.9 mV (n = 12) for hind paw innervating DRG neurons isolated from NC-LV and GRK6-LV groups, respectively. Therefore, GRK6 overexpression significantly hyperpolarized RPs (Figure 4(B); *p < .05 compared with NC-LV, Mann–Whitney test). The AP thresholds were −33.6 ± 2.9 mV (n = 12) and −22.3 ± 1.1 mV (n = 12) for NC-LV and GRK6-LV groups, respectively. GRK6 overexpression remarkably depolarized the AP threshold (Figure 4(C); *p < .01, compared with NC-LV, Mann–Whitney test). The rheobase measurements were 31.7 ± 8.0 (n = 12) and 85.8 ± 12.4 pA (n = 12) for NC-LV and GRK6-LV groups, respectively. GRK6 overexpression also markedly increased rheobase (Figure 4(D); *p < .05, compared with NC-LV, two-sample t test). In addition, GRK6 overexpression resulted in a marked reduction in the numbers of APs evoked by 100 and 300 pA ramp current stimulation (Figure 4(E) to (H); *p < .05 compared with NC-LV, two-sample t test). The numbers of APs evoked by 100 pA ramp current stimulation were 10.4 ± 1.4 (n = 12) and 1.5 ± 0.8 (n = 12) for NC-LV and GRK6-LV groups, respectively. The numbers of AP evoked by 300 pA ramp current stimulation were 14.1 ± 2.2 (n = 12) and 7.0 ± 1.8 (n = 12) for NC-LV and GRK6-LV groups, respectively. GRK6 overexpression significantly decreased the numbers of APs in response to ramp current stimulation.
Reversal of hyperexcitability of DRG neurons in CCI rats by GRK6 overexpression. (A) Bright-field (a), GRK6 LV-GFP (b), DiI fluorescence (c), and merge of GFP and Dil fluorescence (d) images of acutely dissociated DRG neurons. A hind paw innervating DRG neuron infected by GRK-LV is indicated by arrow. Bar = 50 µm. (B) GRK6 overexpression significantly hyperpolarized the RP of hind paw innervating DRG neurons. *p < .05 vs. NC-LV. (C) GRK6 overexpression notably depolarized the AP threshold (*p < .05 vs. NC-LV). (D) GRK6 overexpression also markedly increased rheobase. *p < .05 vs. NC-LV. (E, G) Examples of APs by 100 and 300 pA ramp current injection from NC-LV and GRK6-LV-infected DRG neurons. (F, H) Bar graph showed a significant decrease in numbers of APs evoked by 100 and 300 pA ramp current stimulation by GRK6 overexpression. *p < .05 vs. NC-LV. RP: resting membrane potential; AP: action potential; NC-LV: negative control lentivirus.
Overexpression of GRK6 reduces CXCR2 upregulation
CXC family chemokines have been reported to play a vital role in pain regulation via respective receptors. To determine whether the chemokine receptors are regulated by GRK6 in neuropathic pain, we first detected the change in CXCR1, CXCR2, and CXCR4 protein levels at seven days after CCI application. We selected this time point to perform experiments because both PWT and PWL were at the lowest point on the time-response curve (Figure 1(a) and (b)). As shown in Figure 5(a) to (c), the protein expression of CXCR1, CXCR2, and CXCR4 were all increased markedly in CCI rats when compared with Sham group (*p < .05, two-sample t test). We then investigated whether the upregulation of CXCR1, CXCR2, and CXCR4 was mediated by GRK6. GRK6-LV was used in this study as described above. GRK6 overexpression significantly reversed the CXCR2 upregulation (*p < .05, two-sample t test, Figure 5(e)) but did not alter the expression of CXCR1 (Figure 5(d), p > .05, two–sample t test) and CXCR4 (Figure 5(f), p > .05, two-sample t test).
Reversal of CXCR2 upregulation by GRK6 overexpression in CCI rats. Western blot assays demonstrated a significant upregulation of CXCR1 (a), CXCR2 (b), and CXCR4 (c) protein expression seven days after CCI. *p < .05 vs. Sham. GRK6 overexpression remarkably downregulated CXCR2 protein expression (e) but not CXCR1 (d) and CXCR4 (f) protein expression in CCI rats. *p < .05 vs. NC-LV. CCI: chronic constriction injury; NC-LV: negative control lentivirus.
CXCR2 is associated with GRK6 and CXCR2 inhibitor attenuates CCI-induced neuropathic pain
To further confirm the role of GRK6 on CXCR2 expression in CCI model, we then examined the time course of CXCR2 protein expression and correlation between the expression of CXCR2 and GRK6 of DRGs after CCI. Western blot analysis showed a clear increase in CXCR2 expression persisted from 3 days to 21 days following CCI (Figure 6(a)). As expected, correlation analysis showed that the alternations of CXCR2 and GRK6 following CCI were in a negative correlativity (Figure 6(b), r = 0.824, p = .042). In addition, Co-IP showed an interaction of GRK6 and CXCR2 in DRGs of CCI rats (Figure 6(c)). Immunofluorescence analysis demonstrated that CXCR2 was co-expressed in GRK6 positive DRG neurons (Figure 6(d)). To further verify the function of CXCR2 in neuropathic pain of rats with CCI, we showed that intrathecal injection of SB225002, a selective antagonist of CXCR2, significantly suppressed mechanical allodynia (Figure 6(e), *p < .05 vs. NS, Mann–Whitney test following Friedman ANOVA) and heat hyperalgesia (Figure 6(f), *p < .05 vs. NS, Mann–Whitney test following Friedman ANOVA) in CCI rats.
Correlation of GRK6 and CXCR2 expression and attenuation of neuropathic pain by CXCR2 inhibitor. (a) Expression of CXCR2 protein in DRGs was increased from day 3 to day 21 following CCI. *p < .05 vs. Sham. (b) Correlation analysis showed that the alternations of CXCR2 and GRK6 following CCI were negatively correlated. r2 = 0.824, p = .042. (c) Co-immunoprecipitation showed the co-localization of CXCR2 and GRK6 in DRGs in CCI rats. (d) Immunofluorescence analysis showed CXCR2 was co-expressed in GRK6 positive DRG neurons. GRK6-positive cells (top left) and β-tubulin-positive cells (bottom left) shown in green. CXCR2-positive cells were shown in red (middle column). Merge of double labeling of GRK6 and CXCR2 (top right) and merge of β-tubulin positive staining and CXCR2 labeling (bottom right). Scale bar = 50 µm. (e, f) Administration of the selective CXCR2 antagonist SB225002 mitigated mechanical hyperalgesia and heat hyperalgesia in CCI rats. Antinociceptive effects of a single intrathecal (it.) injection of 20 µg SB225002 were observed at 1 h and disappeared at 2 h. Antinociceptive effects induced by a single injection of SB225002 at 40 µg were observed at 1 h and disappeared 6 h after injection. Injection of SB225002 (10 µg, i.t.) did not alter the PWT and PWL of CCI rats. N = 6 rats for each group, *p < .05, compared to NS group. PWL: paw withdraw latency; PWT: paw mechanical withdrawal thresholds.
Discussion
The present study has shown that GRK6 plays a pivotal role in the maintenance of peripheral sensitization in neuropathic pain via regulation of CXCR2 signaling. Our data showed that CCI produced mechanical allodynia and thermal hyperalgesia accompanied with a significant reduction in GRK6 protein expression, which are located in DRG neurons. Overexpression of GRK6 by lentiviral injection into sciatic nerve significantly attenuated CCI-induced pain responses and markedly reversed hyperexcitability of DRG neurons innervating the sensitized plantar. Furthermore, we demonstrated here for the first time that overexpression of GRK6 suppressed CXCR2 expression in rat DRG. These data suggest that overexpression of GRK6 attenuates mechanical allodynia and thermal hyperalgesia most likely through suppression of CXCR2 expression in rat DRGs.
As reported previously, decreased GRK6 levels led to a markedly decrease in pain threshold.17,18 Female GRK6−/− mice presented enhanced visceral hyperalgesia post-colitis. 17 Mechanical allodynia and thermal hyperalgesia induced by intraplantar injection of inflammatory mediators are also pronounced and prolonged in GRK6−/− mice. 18 Our study showed CCI-induced mechanical allodynia and heat hyperalgesia correlated downregulation of GRK6 protein expression in ipsilateral DRGs, which is in line with the declined GRK6 mRNA expression in ipsilateral DRGs reported recently in another neuropathic pain model of L5 nerve transection. 18 Attenuation of pain responses and reversal of the excitability of sensory neurons by GRK6 overexpression in DRGs indicated the peripheral role of GRK6 in neuropathic pain, which was consisted with our previous report about the expression and role of GRK6 in spinal cord dorsal horn of CCI rats. 19
The mechanism underling negative regulation of GRK6 on pain hypersensitivity is largely unknown. GRK6, as a ubiquitously expressed member of the GRK family, has been described to experience a marked change and play a critical role in inflammatory disease in humans or animal models. Profound downregulation of GRK6 expressions have been reported in rheumatoid arthritis or multiple sclerosis.11,12 Interestingly, the data of the previously published pain-related behavior tests in GRK6-/- and WT animals showed that at baseline there was no difference in pain threshold between these two groups. This indicated that low GRK6 level per se is not sufficient to decrease pain threshold except participation of an inflammatory stimulus. Therefore, we speculated that the role of decreased GRK6 in DRGs might be associated with the neuroinflammation. There is growing body of evidence that chemokines and their receptors play a role in inducing and maintaining neuropathic pain via GPCRs. GRK6 and other members of the GRK family have been originally identified because of their capacity to phosphorylate agonist-occupied GPCRs and regulate GPCR desensitization and internalization. Of high interest, the receptors of CXC chemokine family, the cysteine residues are separated by any amino acid residue, have been revealed associated with GRK6 implicating in several pathophysiological conditions.15,30 Our data showed that the expressions of CXCR1, CXCR2, and CXCR4 are all upregulated in DRGs of CCI rats, which is consisted with recent studies about the receptors of CXC chemokine family in pain regulation. It has been reported that CXCR2 predominantly interacts with GRK6, whereas CXCR1 couples to GRK2 to negatively regulate receptor sensitization and trafficking in cell signaling. 30 In addition, site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. 36 Our finding showed that only the increased CXCR2 was reversed by GRR6 overexpression in CCI rats, which indicated that GRK6-induced antinociceptive role might be mediated by inhibition of CXCR2 expression in DRG neurons. This conclusion is based on the following observations. Firstly, overexpression of GRK6 reversed the increased level of CXCR2 without altering the expression of CXCR1 and CXCR4 in DRG of CCI rats. Secondly, the temporal expression of GRK6 and CXCR2 in CCI rats is negatively correlated. Thirdly, Co-IP showed the interaction of CXCR2 and GRK6 in DRGs of CCI rats. Fourthly, co-immunofluorescence analysis demonstrated that CXCR2 co-expressed on GRK6 positive DRG neurons. Finally, inhibition of CXCR2 by SB225002 suppressed the pain hyperalgesia induced by CCI. Although more experiments are required to investigate whether the GRK6-mediated CXCR2 desensitization is involved in neuropathic pain regulation, we showed for the first time the mechanism of GRK6 in pain regulation via GPCR (i.e. CXCR2) signaling under neuropathic conditions. Of note, roles of CXCR1 and CXCR4 in CCI rats can not be excluded although the expression of CXCR1 and CXCR4 was not affected by GRK6 overexpression. However, the expression of CXCR1 and CXCR4 was also increased in CCI rats. Previous studies have reported that AMD3100, a potent and selective CXCR4 inhibitor, was proved to reverse the BV-induced persistent pain, 28 and that DF2755, a novel non-competitive inhibitor of CXCR1/2, reduced inflammatory and post-operative pain. 27 Considering these, we proposed that CXCR1 and CXCR4 are likely to modulate neuropathic pain through GRK6-independent signal pathways, which definitely deserves to be investigated in the future.
More interestingly, GRK6 overexpression via lentivirus treatment also reversed the hyperexcitability of DRG neurons from CCI rats, which were demonstrated by a significant hyperpolarization of the RP, a depolarization of action membrane threshold, an increase in rheobase, and a reduction in the number of APs evoked by the injection of different sizes of current ramps. CXCL1, the primary ligand of CXCR2, have been revealed modulating neuronal excitability of DRG neurons.37–39 CXCR2 is a GPCR that has been shown to activate a number of different signaling pathways in neurons, including intracellular Ca2+, inositol tris-phosphate, MAP kinases, and CREB, thus leading to modulate ion channels and excitability. 40 Our explanation for reversal of excitability by GRK6 overexpression is GRK6-mediated CXCR2 desensitization, which is consistent with the results illustrated in present study. However, we cannot entirely rule out the possibilities that GRK6 may impact other GPCRs involved in hyperalgesia, and that GRK6 also can bind and phosphorylates other regulators of ion channels (such as PDZ domains in Na+/H+ exchanger regulatory factor and downstream regulatory element antagonistic modulator) via mechanism independent of GPCR desensitization to regulate hyperalgesia.10,40 Synaptic plasticity, as a pivotal mechanism for chronic pain, also occurs at different levels of the central nervous system. Nerve injury triggers long-term plastic changes along sensory pathways, from the peripheral sensory terminals to the spinal cord dorsal horn, 41 amygdale, 42 and anterior cingulate cortex.43–45 In addition, increasing literature implicated a role of GRK6 in central sensitization of neuropathic pain. GRK6 has been reported expressed in neurons of spinal cord, 19 cortex, 46 and hippocampus. 47 Whether GRK6 is involved in the maintenance of pain-related long-term synaptic plasticity changes in spinal and/or supraspinal synapses needs further exploration. It is worth to be mentioned here, that the method of lentivirus injection through sciatic nerve, different from traditional intrathecal injection, has profound advantage in demonstrating the peripheral mechanism of neuropathic pain. We showed that transgene expression was successfully achieved in the sciatic nerves and the corresponding DRG but not in other irrelative DRGs and spinal cord. It is clear and definite to illustrate the peripheral role of the transgene in pain modulation. In addition, this injection is controllable and convenient to be conducted with negligible nerve damage compared with nerve injury of neuropathic pain model. Future experiments are warranted to optimize the approaches for LV delivery into DRGs.
In conclusion, to the best of our knowledge, we provide the first demonstration that GRK6 plays a pivotal role in the maintenance of peripheral sensitization in neuropathic pain via regulation of CXCR2 signaling pathway. The present results and future studies might identify a potential therapeutic target for the treatment of the intractable neuropathic pain.
Footnotes
Author Contributions
YZ researched, analyzed data, and wrote the manuscript. RJL researched, analyzed data, and wrote the manuscript. XL researched and analyzed data. HYZ researched data. ML researched and analyzed data. ZJ analyzed data. XM analyzed data and wrote the manuscript. GYX designed and supervised the experiments, and edited the manuscript. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by grants from National Natural Science Foundation of China (81230024 and 81471137) and from the Priority Academic Program Development of Jiangsu Higher Education Institutions of China. This project is also subject to the Second-Affiliated Hospital of Soochow University Preponderant Clinic Discipline Group Project Funding (XKQ2015008). The all funders had no role in the experimental designs, data collection and analyses, decision to publish, or preparation of the manuscript.