
Abstract
Mitochondria, known as the powerhouses of cells, are considered a key source of reactive oxygen species (ROS) production in various cell types. In the context of neuropathic and inflammatory pain, both mitochondrial dysfunction and hyperfunction can lead to aberrant production of mitochondrial reactive oxygen species (mtROS), which has been implicated in the development and persistence of pain hyperalgesia. This comprehensive review delves into the compelling correlation between mitochondrial functional activity and diverse pain conditions, with a special emphasis on inflammatory pain and chemotherapy-induced peripheral neuropathy (CIPN). Furthermore, it explores the therapeutic potential of targeting mitochondrial protection and mtROS scavenging to maintain mitochondrial redox homeostasis, offering a novel approach for pain management. The findings presented here provide valuable insights into the multifaceted role of mitochondria in pain modulation, laying a solid foundation for future research and the development of innovative analgesic strategies.
Introduction
Mitochondria, commonly known as ‘the powerhouse of the cell’, are indispensable for maintaining normal cellular function and promoting overall human health. They play a vital role in a wide range of critical tasks, encompassing energy production, the electron transport chain, cell signaling, and apoptosis. These functions are essential for regulating various biological processes that are necessary for cell survival and precise functioning. 1 The distribution of mitochondria is tailored to meet the varied adenosine triphosphate (ATP) requirements of different cells. Notably, sensory neurons, which have high energy demands, tend to concentrate mitochondria. 2 Consequently, any disruption or malfunction in these crucial processes can have profound consequences on health. Research has demonstrated the involvement of mitochondrial dysfunction and the production of mtROS as key factors in the development of neurodegenerative disorders like Alzheimer's disease and Parkinson's disease. They play a central role in the pathological processes underlying these conditions.3,4 Furthermore, emerging evidence suggests a strong correlation between mitochondrial dysfunction/hyperfunction and diverse pain conditions, including inflammatory pain and neuropathic pain.5,6
Approximately 90% of cellular ROS are produced by mitochondria, primarily via oxidative phosphorylation (OXPHOS) and the mitochondrial electron transport chain (mETC) (Figure 1). The key contributors to mtROS production are mitochondrial complexes I and III, predominantly located within the respiratory chain. 7 These unpaired electrons then react with O2, resulting in the production of highly reactive free radicals known as superoxide ions. These superoxide ions can further be converted into other mtROS, such as H2O2 and hydroxyl ions (−OH). 8 Under normal conditions, a balance between mtROS production and elimination is maintained. However, under pathological stress, mitochondrial dysfunction/hyperfunction can contribute to an excessive generation of mtROS. Structurally compromised mitochondria become net mtROS producers due to impaired removal systems, such as the glutathione peroxidase/glutathione reductase cycle, 9 while hyperfunctional mitochondria stimulate an increase in mtROS production through enhanced mitochondrial respiration. 10 Both contribute to oxidative stress, which may play a significant role in the development of inflammatory and neuropathic pain.

mtROS Production via OXPHOS and the Mitochondrial Electron Transport Chain (mETC).
In this review, we will focus on the existing knowledge underlying the role of mitochondrial function and mtROS homeostasis in the maintenance and development of inflammatory and neuropathic pain. Our objective is to provide valuable insights into potential molecular targets that can be explored as clinical treatments for these conditions.
Inflammatory pain
Dysfunctional mitochondria contribute to substantial mtROS generation in the context of inflammatory pain
Mitochondria and mtROS play significant roles in the development and maintenance of pain hyperalgesia, particularly through central sensitization in inflammatory pain (Figure 2). The activation of the Transient Receptor Potential Vanilloid type 1 (TRPV1) channel by capsaicin triggers inflammatory pain. 11 This model exhibits two distinct pain components: primary hyperalgesia, associated with peripheral sensitization, and secondary hyperalgesia, reflecting central sensitization. In experiments conducted on normal mice injected with capsaicin into the left hind foot, both primary and secondary mechanical hyperalgesia develop and persist for 24 h. Administration of Phenyl-N-tert-butylnitrone (PBN), a non-specific ROS scavenger, or 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPOL), a superoxide selective scavenger, systemically or intrathecally, results in a dose-dependent reduction of secondary hyperalgesia only, while primary hyperalgesia is largely unaffected. These findings suggest that mtROS play a crucial role in the development and persistence of capsaicin-induced hyperalgesia in mice, primarily through central sensitization. 12 Moreover, mitochondrial superoxide is a major contributor to elevated spinal mtROS levels in capsaicin-induced hyperalgesia. 13 Mitochondrial superoxide may therefore serve as a potential target for reducing central sensitization.

The effects of mitochondrial hyperfunction/hypofunction on neurons.
Various pain models, such as acute inflammatory pain induced by formalin and persistent pain induced by Complete Freund's Adjuvant (CFA), have revealed an increased presence of clustered mitochondria and altered mitochondrial distribution in the spinal dorsal horn. Morphological analyses reveal marked differences between physiological and pathological conditions: spinal dorsal horn neurons in healthy mice display mitochondria with dispersed granular distribution, while pain models show characteristic mitochondrial clustering. However, the origin of these increased mitochondria remains unidentified. Notably, neurons undergoing central sensitization exhibit an accumulation of mitochondria, suggesting that aberrant mitochondrial distribution and dysfunction play a role in pain hypersensitivity by promoting central sensitization through extensive mtROS generation. 14 Furthermore, Additional evidence shows that intense nociceptive input leads to heightened production of mitochondrial superoxide, by either enhancing mitochondrial OXPHOS or compromising mitochondrial efficiency due to excessive Ca2+ accumulation. Consequently, the elevated levels of mtROS in the spinal cord sensitize neurons in the dorsal horn, leading to pain perception. 15
Mitochondrial hyperfunction and mtROS in inflammatory pain
Emerging evidence suggests that increased mitochondrial function contributes to the development of inflammatory hyperalgesia. Prostaglandin E2 (PGE2), a pro-inflammatory mediator, enhances mitochondrial pyruvate dehydrogenase (PDHA1) phosphorylation in dorsal root ganglia (DRG) neurons via the EPAC2-PKC signaling pathway. Phosphorylated PDHA1 irreversibly converts pyruvate derived from cytoplasmic glucose into acetyl-CoA, coupling glycolysis to mitochondrial respiration. This acetyl-CoA fuels the tricarboxylic acid cycle, generating reduced electron carriers (NADH and FADH2) that donate electrons to complexes I and II of the electron transport chain (ETC). The resultant proton gradient drives ATP synthesis, amplifying mitochondrial activity. Critically, heightened ETC flux increases electron leakage, particularly at complexes I and III, which directly elevates mtROS production. 16
Recent studies have identified Recombinant Mouse ATP synthase subunit C lysine N-methyltransferase (ATPSc-KMT) as a crucial mitochondrial protein involved in chronic inflammatory pain development. ATPSc-KMT is now defined as the lysine methyltransferase responsible for post-translational modification of mitochondrial ATP synthase. Methylated ATPSc constitutes an essential component for mitochondrial ATP production (Figure 1). Overexpression of ATPSc-KMT leads to elevated mtROS production and is crucial for sustaining efficient mitochondrial respiration. 17 In a λ-carrageenan-induced pain model, both mRNA and protein levels of ATPSc-KMT were found to be increased in DRG neurons. These changes persisted even after the resolution of inflammatory hyperalgesia, suggesting that ATPSc-KMT upregulation in DRG neurons may cause mitochondrial hyperfunction, metabolic disturbances, and hyperalgesia priming. However, whether inhibiting ATPSc-KMT expression can alleviate pain remains to be determined. Moreover, persistent inflammatory pain can be relieved by reducing mitochondrial overactivation, scavenging mtROS, or supplementing nicotinamide adenine dinucleotide (NAD+). 18 NAD+ has emerged as a critical cofactor that regulates mitochondrial fitness and various redox reactions. NAD+ supplementation can mitigate enhanced glycolysis and redox imbalances.19,20
Additionally, it has been observed that M2 macrophages can effectively and transiently resolve λ-carrageenan-induced inflammatory pain by transferring mitochondria to sensory neurons. This transfer is accompanied by the restoration of OXPHOS, indicating that the transferred mitochondria likely replace damaged ones and restore mitochondrial homeostasis in sensory neurons. 21 These findings highlight the involvement of mitochondria in the endogenous mechanisms responsible for actively resolving inflammatory pain and emphasize the importance of maintaining precise regulation of mitochondrial and metabolic activity to facilitate the resolution of inflammatory pain. This also suggests a novel strategy for pain relief: artificially increasing exogenous healthy M2 cells and using them to transfer healthy mitochondria into cells with abnormal mitochondrial function.
Neuropathic pain
Mitochondrial dysfunction and mtROS in neuropathic pain
Extensive evidence has demonstrated the critical role of dysfunctional mitochondria and mtROS in the development of neuropathic pain. In an L5 spinal nerve ligation model of neuropathic pain, it was observed that ROS generation primarily occurs in the mitochondria of dorsal horn neurons. 22 Similarly, altered mitochondrial distribution and increased mitochondrial clustering were detected within the spinal dorsal horn in a model of neuropathic pain induced by spared nerve injury (SNI). 14 Mitochondria have also been implicated as a major source of ROS in cancer-induced bone pain (CIBP). 23 Furthermore, findings from a spinal cord injury-induced neuropathic pain model suggest that dysfunctional mitochondria contribute to elevated cytosolic calcium levels by promoting superoxide production and impairing calcium uptake in neurons. This, in turn, activates calcium-calmodulin–dependent protein kinase II (CaMKII) and promotes L-type calcium channel activity, ultimately leading to enhanced neuronal excitation and synaptic transmission, leading to mechanical allodynia. 24 The role of mitochondrial function in the onset and persistence of neuropathic pain has been extensively studied, particularly in chemotherapy-induced animal models.
Classical mitochondrial dysfunction and mtROS in CIPN
Chemotherapy-induced formation of atypical mitochondria
CIPN, characterized by neuropathic pain, is closely associated with abnormal and dysfunctional mitochondria.25,26 An electron microscopy study has demonstrated the presence of swollen and vacuolated mitochondria in both C-fibers and myelinated axons of nerves treated with paclitaxel. 27 These atypical mitochondria in neurons may contribute to the development and persistence of paclitaxel-induced pain syndrome, which manifests before and during pain episodes but disappears once the syndrome resolves.2,28 Swollen and vacuolated mitochondria have also been observed in both C-fibers and A-fibers of rat models experiencing painful neuropathy induced by oxaliplatin29,30 and bortezomib. 31 Current evidence reveals distinct chemotherapy-specific effects on Schwann cell mitochondria. For example, cisplatin and carboplatin directly impair mitochondrial function in Schwann cells, triggering demyelination and apoptosis, while oxaliplatin spares mitochondria and lacks comparable neurotoxic effects. 32 However, overall, chemotherapeutic agents tend to promote mitochondrial-mediated demyelination and apoptosis in Schwann cells. 33 However, none of these drugs, including paclitaxel, oxaliplatin, and bortezomib, were found to induce atypical mitochondria in Schwann cells. Observations of mitochondrial changes in Schwann cells contradict the conclusions reported in most studies.
Dysfunction of atypical mitochondria
Previous studies have shown that swollen and vacuolated mitochondria in peripheral nerves impair mitochondrial respiration and ATP production. In the case of paclitaxel-induced and oxaliplatin-induced peripheral neuropathy, affected nerves exhibited significantly reduced rates of stimulated respiration for both Complex I-mediated and Complex II-mediated processes, leading to a notable decrease in stimulated ATP production. 29 Moreover, bortezomib and paclitaxel have been found to promote glycolysis while suppressing OXPHOS in sensory neurons, despite the fact that glycolysis is less efficient in generating ATP compared to OXPHOS.34,35 Moreover, bortezomib can deplete mitochondrial NAD+ through two mechanisms: first, by inducing the degradation of nicotinamide mononucleotide adenylyltransferase 2, a key enzyme in NAD+ biosynthesis that catalyzes the conversion of nicotinamide mononucleotide to NAD+; second, by activating sterile alpha and TIR motif-containing 1, an NAD+-degrading enzyme. This dual action ultimately impairs ATP synthesis. The effects of three drugs on mitochondrial ATP production and their potential mechanisms were summarized in Table 1. Impaired mitochondrial ATP production in peripheral sensory neurons is considered a potential mechanism underlying CIPN. However, the administration of glucose, which would enhance both glycolytic and mitochondrial ATP production, did not alleviate pain in mice treated with bortezomib. This suggests that CIPN-associated pain may be driven by the accumulation of metabolites from aerobic glycolysis. 34
Effects and mechanisms of chemotherapeutic agents on mitochondrial ATP generation.

Induction of aberrant mtROS by dysfunction mitochondria
mtROS also play a pivotal role in both the development and maintenance of CIPN. Extensive research has investigated the impact of oxidative stress on CIPN using antioxidants. 37 For example, PBN has successfully reduced mechanical hypersensitivity in mice treated with cisplatin and paclitaxel.38–40 Antioxidants like N-acetylcysteine, alpha-lipoic acid, and vitamin E have demonstrated the potential to prevent and reverse Oxaliplatin-induced mechanical hypersensitivity. This effect is attributed to their ability to inhibit mtROS production, alleviate oxidative damage, and suppress pro-inflammatory signals in the spinal cord. 41 Mitoquinone has been discovered to alleviate vincristine-induced neuropathic pain by inhibiting oxidative stress and apoptosis through enhanced mitochondrial function. 42 Mannose-coated superparamagnetic iron oxide nanoparticles (mSPIONs) have also exhibited strong anti-ROS and inflammation-alleviating effects, effectively relieving vincristine-induced CIPN. 43 Notably, while mitochondrial dysfunction can increase mtROS production, ROS can further exacerbate mitochondrial damage. Furthermore, it is yet to be determined if there are other cellular sources of ROS contributing to CIPN. The exact origin of ROS and the complex interplay between ROS and mitochondrial dysfunction in the context of CIPN require further investigation. Additionally, it remains unclear whether mitochondrial hyperfunction occurs in CIPN and if elevated mtROS levels resulting from hyperfunction contribute to its onset and progression. Further research is needed to clarify these mechanisms.
Therapies aimed at reducing mtROS levels
As previously discussed, the excessive and prolonged increase in mtROS production and oxidative stress is a key factor in the development of neuropathic pain. This underscores the potential of antioxidants as a therapeutic strategy for pain management.
Antioxidant
Acetyl-L-carnitine (ALC) functions as an antioxidant and plays a crucial role in the metabolism of free fatty acids. Preemptive administration of ALC has shown promise in protecting against mitochondrial dysfunction induced by oxaliplatin, paclitaxel, and bortezomib, while also reducing the onset of neuropathic pain.30,31 Although clinical studies support the effectiveness of ALC in preventing and alleviating paclitaxel-induced pain,44,45 some evidence suggests that prolonged ALC treatment may worsen CIPN. 46 However, this study represents a clinical trial currently lacking supporting fundamental experimental evidence. The potential mechanisms by which long-term ALC use may paradoxically exacerbate CIPN-associated pain remain unclear and need more studies. Furthermore, vitamin C (a ubiquitous antioxidant) shows an inverse correlation with postoperative analgesic demand (r = −0.699, p < 0.001) in orthognathic surgery patients, suggesting its role in pain modulation. 47
Not only conventional Western drugs but also numerous traditional Chinese medicines (TCM) exhibit notable anti-inflammatory and antioxidant properties, with emerging clinical evidence supporting their therapeutic potential. For instance, preclinical and clinical studies have shown that curcumin activates the ERK1/2 signaling pathway, thereby conferring neuroprotective effects in neurodegenerative models. Similarly, matrine has been validated in clinical trials to exhibit antioxidant, anti-inflammatory, and calcium-antagonizing properties, effectively alleviating neuropathic pain with favorable safety profiles. 48 Notably, a meta-analysis incorporating 57 clinical studies demonstrated that TCM interventions achieved superior efficacy, more significant analgesic effects, and fewer adverse reactions compared to conventional Western medicine in treating low back pain, underscoring their clinical value. 49 However, current antioxidant agents lack precise targeting capabilities at cellular or subcellular levels for effective ROS clearance, leading to limited therapeutic efficacy. Additionally, some antioxidants may disrupt normal redox signaling pathways while failing to reverse pre-existing oxidative damage. 50
Ozone (O3)
O3 is a gas known for its medical applications due to its antioxidative and anti-inflammatory properties. In a rat model of streptozotocin-induced diabetic neuropathy, ozone exhibited preventive effects by modulating oxidative stress and antioxidant defenses. 51 Additionally, ozone influences the inflammatory response by reducing pain signaling pathways. 52 These findings suggest that ozone may target multiple mechanisms involved in CIPN development, making it a potential adjunctive therapy for CIPN treatment. 53 In cervical pain treatment, paravertebral muscle injections of oxygen-ozone mixtures have demonstrated both efficacy and safety. 54 For fibromyalgia management, ozone autohemotherapy significantly reduces patients' visual analog scale scores and Fibromyalgia Impact Questionnaire ratings while improving quality of life, with benefits sustained for three months. 55 A double-blind trial on knee osteoarthritis revealed that intra-articular ozone injections at 20 µg/ml and 40 µg/ml concentrations effectively alleviated pain and enhanced functional mobility in patients. Similarly, 56 in temporomandibular joint osteoarthritis, ozone-treated patients showed significantly better masticatory efficiency and pain relief compared to intra-articular anesthesia alone. 57 However, beyond its anti-inflammatory and antioxidant properties, ozone exhibits intrinsic irritancy. For instance, high-concentration ozone injections may cause transient burning sensations at administration sites during joint pain therapy, 58 limiting its clinical utility. Therefore, in clinical practice, ozone parameters (concentration and combination protocols) must be optimized according to disease type (e.g., neuropathic vs musculoskeletal pain) to balance therapeutic efficacy and safety.
Photobiomodulation (PBM)
PBM, which is a therapeutic technique that utilizes specific wavelengths of light (e.g. red light at 620–700 nm and near-infrared light at 700–1440 nm) to deliver non-invasive bio-stimulatory effects on cells and tissues, its mechanisms include anti-inflammatory action, tissue repair promotion, and analgesia. It is worth noting that although low levels of light have stimulating effects, high levels of light can produce inhibitory effects. Studies demonstrate that PBM enhances mitochondrial function by activating cytochrome c oxidase (CCO), which elevates mitochondrial membrane potential (MMP) and improves ETC efficiency. In healthy cells, this process induces minimal electron leakage from the ETC, generating transient superoxide, and low level ROS. Conversely, in oxidative-stressed cells or disease models, mitochondrial dysfunction causes excessive electron leakage, leading to pathological ROS accumulation. PBM restores CCO activity, normalizes MMP, and mitigates electron leakage, thereby reducing ROS production to physiological levels.
Clinically, PBM has been widely applied for analgesia in dental patients, particularly in managing chemotherapy-induced oral mucositis and post-surgical dental pain.59,60 For example, a triple-blind trial on fibromyalgia treatment demonstrated that PBM therapy significantly improved pain severity, functional capacity, and psychological symptoms.61,62 Additionally, PBM exhibits notable efficacy in neuropathic pain management, as it enhances nerve conduction velocity and alleviates pain in clinical cases of spinal cord injury or diabetic neuropathy. 63 By enhancing antioxidant defenses and mitigating oxidative stress, PBM presents a promising treatment option for inflammatory pain.64,65
Although PBM is generally considered a safe therapeutic approach, potential risks may still exist under certain circumstances. For instance, high laser intensities may induce pain or discomfort and could even cause tissue damage. 66 Additionally, its therapeutic efficacy varies across different pain types, and PBM is not universally effective for all pain conditions. For example, while most studies demonstrate significant analgesic effects of PBM in managing oral mucositis pain, its efficacy may be limited in severe mucositis cases or those with complications. 67
Drp1 Inhibitors
Dynamin-related protein 1 (Drp1)-mediated mitochondrial fragmentation leads to excessive mtROS production, disrupting mitochondrial redox balance and contributing to neuropathy and neuropathic pain. This imbalance triggers oxidative stress, alters mitochondrial dynamics, and perpetuates mtROS generation. The increased production of mtROS, in turn, further impacts mitochondrial dynamics and exacerbates mtROS generation, forming a vicious cycle involving Drp1, mitochondria, and mtROS. 68 Mitochondrial division inhibitor (Mdivi-1), a selective Drp1 inhibitor, has shown significant potential in inhibiting mtROS generation (Figure 3) and reducing the severity of mechanical hyperalgesia induced by hydrogen peroxide. 69 Therefore, strategies aimed at inhibiting Drp1-induced disorders may offer a novel therapeutic strategy for treating neuropathic pain. However, current small-molecule Drp1 inhibitors face challenges in specificity. For example, while Mdivi-1 is the most widely studied Drp1 inhibitor, its selectivity for human Drp1 remains unconfirmed, and significant off-target effects have been reported in preclinical studies. 70
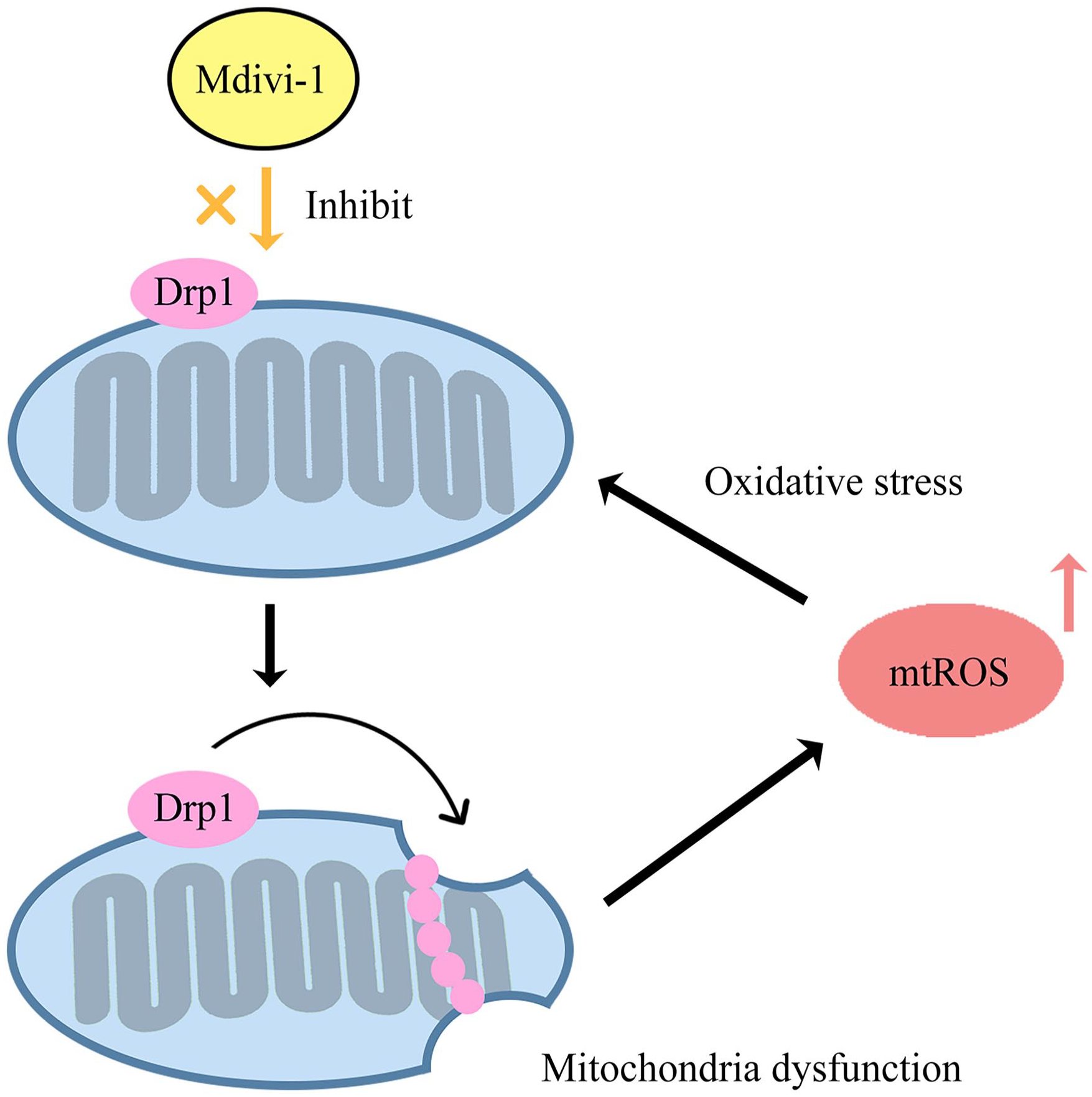
The process of Mdivi-1 promoting mitochondria to generate mtROS.
Quercetin
Quercetin, a bioactive phenolic compound found in blueberry leaves, exhibits strong antioxidant properties. 71 Studies have indicated that quercetin can alleviate osteoarthritis by diminishing the generation of mtROS and augmenting the expression of glutathione (GSH) and glutathione peroxidase (GPx) in osteoarthritis models. Furthermore, quercetin not only enhances mitochondrial membrane potential, oxygen consumption, and ATP levels but also increases the number of mitochondrial copies, effectively reversing mitochondrial dysfunction. 72 However, its precise mechanisms of action require further investigation.
Discussion and prospects
Emerging evidence indicates the significant role of mitochondria and mtROS in the development and persistence of pain hyperalgesia, primarily through central sensitization. Both mitochondrial dysfunction and hyperfunction can induce pain by increasing mtROS levels. Additionally, antioxidants, PBM, and Drp1 inhibitors have shown promise in alleviating mechanical hyperalgesia. However, there are still numerous unanswered questions concerning the modulation and impairment of mitochondrial function in inflammatory and neuropathic pain conditions. Further research is needed to unravel these complex mechanisms and facilitate the development of effective analgesic therapies. Moreover, the effectiveness of most treatments has been demonstrated only in animal models, with limited supporting clinical evidence.
Considering the potential role of mitochondria in the endogenous mechanisms that actively alleviate inflammatory pain, maintaining a balance between mitochondrial production and clearance within cells could be a critical therapeutic target for pain management. Prioritizing this balance holds significant promise for effectively treating and alleviating pain conditions. Pain mitigation can be achieved through various approaches that enhance mitochondrial function. Mitochondrial protectors, for instance, primarily improve mitochondrial integrity by promoting biogenesis, increasing mitochondrial quantity and functionality, thereby boosting cellular energy metabolism and stress resistance. Meanwhile, gene editing-a highly promising therapeutic strategy, can also be applied to optimize mitochondrial performance. One approach involves engineering healthy macrophages and transplanting them into damaged tissues, enabling the transfer of functional mitochondria to cells with impaired mitochondrial activity.
CRISPR-based gene editing has advanced significantly technically, it remains suboptimal for modifying mitochondrial genes due to the inefficient delivery of RNA into mitochondria. Key challenges persist, including off-target effects, ethical concerns, and delivery limitations for clinical translation.73–75 Meanwhile, non-CRISPR methods (e.g., engineered nucleases and deaminases) have shown progress in facilitating mtDNA disruption or base editing. 73 While not yet widely adopted in preclinical studies, these innovations provide a conceptual foundation for future gene-editing therapies for CIPN.
Footnotes
Appendix
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (81400916 to Dr X. Zhu), Natural Science Foundation of Hunan Province (2021JJ41060, 2025JJ50532 to Dr X. Zhu), Research Program Project of Hunan Health Commission (B202304119634 to Dr X. Zhu).