
Abstract
Botulinum neurotoxins (BoNTs), produced by Clostridium botulinum, have been used for the treatment of various central and peripheral neurological conditions. Recent studies have suggested that BoNTs may also have a beneficial effect on pain conditions. It has been hypothesized that one of the mechanisms underlying BoNTs' analgesic effects is the inhibition of pain-related receptors' transmission to the neuronal cell membrane. BoNT application disrupts the integration of synaptic vesicles with the cellular membrane, which is responsible for transporting various receptors, including pain receptors such as TRP channels, calcium channels, sodium channels, purinergic receptors, neurokinin-1 receptors, and glutamate receptors. BoNT also modulates the opioidergic system and the GABAergic system, both of which are involved in the pain process. Understanding the cellular and molecular mechanisms underlying these effects can provide valuable insights for the development of novel therapeutic approaches for pain management. This review aims to summarize the experimental evidence of the analgesic functions of BoNTs and discuss the cellular and molecular mechanisms by which they can act on pain conditions by inhibiting the transmission of pain-related receptors.
Introduction
The International Association for the Study of Pain (IASP) defines pain as an “An unpleasant sensory and emotional experience associated with actual or potential tissue damage”. 1 Pain is a common reason for seeking medical care, with osteoarthritis, back pain, and headaches ranking among the top 10 causes.2,3 It can be classified as acute or chronic based on the duration of symptoms, with chronic pain persisting or recurring for more than 3 months. 4 Furthermore, pain can be classified into three primary types based on its underlying causes or pathogenesis: nociceptive or inflammatory pain, neuropathic pain, and mixed pain.5–7 Despite advancements in understanding the neurobiological mechanisms of pain, its management remains unsatisfactory, with less than 50% of patients finding relief from initial medications.8,9 There is a need for more effective analgesics, which requires a deeper understanding of pain mechanisms. Neuropathic pain is a chronic condition that poses challenges in clinical treatment due to its limited response to therapies compared to other types of pain.10,11 It is associated with an imbalance between excitatory and inhibitory somatosensory signaling, as well as variations in the processing of pain signals within the central nervous system. The condition is resistant to conventional treatment approaches, and its management can be particularly difficult. 12 The two main symptoms of neuropathic pain, allodynia and hyperalgesia, occur due to the release of inflammatory mediators, which act on nociceptive nerve endings, lowering the threshold for neuronal excitation. This can lead to a phenomenon known as central sensitization.13,14 In cases where the somatosensory nervous system is damaged, the release of inflammatory mediators can trigger ectopic discharge of nerve fibers. This abnormal firing of nerves results in neuronal hyperexcitability, thereby causing symptoms such as hyperalgesia, allodynia, and spontaneous pain.10,15,16 Neuropathy can disrupt the sensory signaling pathway in both the spinal cord and brain by affecting the expression and function of ion channels and receptors within neurons. Numerous ion channels and receptor classes have been identified as potential targets for the development of next-generation analgesic drugs. These channels include potassium channels, sodium channels, ATP-gated channels, transient receptor potential channels, and calcium channels.17–19 The receptor classes include purinergic receptors, the neurokinin-1 receptor, AMPA receptors, NMDA receptors, glutamate receptors, and opioid receptors. These receptors contribute to different levels of pain signaling and are potential targets for therapeutic interventions.20–23
Botulinum neurotoxin (BoNT) is a potent neurotoxin used to address pain, particularly neuropathic pain, and has been applied in various medical treatments for disorders like dystonia and blepharospasm.12,24 Numerous clinical trials, case studies, and animal models have demonstrated BoNT’s efficacy in managing neuropathic pain. However, the mechanisms underlying BoNT’s role in pain signaling are subject to debate. Growing evidence suggests that BoNT’s analgesic and anti-inflammatory effects are mediated through multiple mechanisms, indicating a complex interplay in its pain-relieving actions. BoNT therapy alleviated pain through two primary mechanisms. Firstly, it directly reduces muscle contraction by inhibiting acetylcholine release, leading to relief from pain. Secondly, it exerts indirect effects by inhibiting the release of neurotransmitters other than acetylcholine and modulating receptors and ion channels involved in pain signaling. These combined actions
In this review, we aim to provide a comprehensive summary of the potential effects of botulinum neurotoxin on receptors associated with pain. By exploring these mechanisms, we can gain a better understanding of how BoNT exerts its analgesic effects.
Botulinum neurotoxin
BoNTs are a class of bacterial proteins produced by the anaerobic bacteria “Clostridium botulinum”. These toxins exhibit potent inhibitory effects on synaptic transmission within the peripheral cholinergic nervous system. 25 There are seven distinct serologically different BoNT isoforms, designated as A-G [40], and each subtype of BoNT has a different amino acid sequence. 26 Human botulism, a neuroparalytic disease, is primarily caused by four serotypes of BoNT: A, B, E, and F. Types C and D are responsible for inducing botulism in domestic and wild animals but are not known to affect humans. A bacterial species that produces the toxin type G was discovered in South American soil in 1969. However, it has never been associated with causing foodborne botulism.27–29 In recent years, significant progress has been made in understanding the structures and mechanisms of action of BoNTs. These toxins exhibit neurospecificity, targeting peripheral nerve terminals associated with skeletal and autonomic cholinergic nerves, allowing their use as therapeutic agents in treating various human diseases.30,31 FDA-approved BoNT types A and B have been used for conditions such as migraine, hypersalivation, dystonia, overactive bladder, spasticity, strabismus, blepharospasm, and cosmetic procedures.32–38 The duration of BoNT effects varies depending on the serotype, with BoNT/A and BoNT/C typically exhibiting longer-lasting effects (around 4–6 months).39,40
Structure of botulinum neurotoxin
BoNT serotypes consist of a metalloprotease light chain (50 kDa) and a heavy chain (100 kDa) connected by an interchain disulfide bond. The toxin molecule of BoNTs is often enveloped and protected by a group of naturally occurring proteins, in addition to the two peptide chains. These neurotoxins exist as macromolecular complexes ranging in size from 300 kDa to 900 kDa, and the specific molecular weight and structure of the protein complex are determined by the clostridial strain and serotype involved. The 900 kDa complex is unique to serotype A.
28
BoNT serotypes have three functional domains: binding, translocation, and catalytic. The C-terminal of the heavy chain binds the toxin to the receptor site, while the N-terminal of the heavy chain facilitates translocation, while the belt region of the translocation domain contributes to the protection of the active site. The BoNT light chain serves as the catalytic domain and functions as a zinc-dependent endopeptidase. These domains are arranged linearly, with the translocation domain located in the center. Physicochemical tests have shown that each clostridial neurotoxic molecule, except for BoNT/C, contains one zinc atom linked to the light chain. BoNT/C, however, contains two zinc atoms.41,42 The receptor-binding domain of the heavy chain of BoNT initially interacts with polysialogangliosides on the presynaptic surface, and then the toxin binds to another surface receptor, such as synaptotagmin (Syt) or synaptic vesicle protein 2 (Sv2), leading to internalization. BoNT serotypes A, D, E, F predominantly bind with Sv2, while serotypes B and G exhibit a strong affinity for Syt.43–45 BoNT serotype C is unique as it has not yet been associated with a specific protein receptor in neuronal cells. Instead, BoNT-C has been observed to bind to liposomes containing phosphoinositide (Figure 1).
46
Mechanism of action of BoNT A and B at neuromuscular junction. 1) BoNT uses the C-terminal of its heavy chain (binding domain) to bind to PSGs on the presynaptic surface. 2) BoNT/A binds to SV2, while BoNT/B binds to the Syt protein. Subsequently, the toxin enters the cell through receptor-mediated endocytosis. 3) The entry of H+ ions into the vesicle through proton pumps causes acidification. As a result, the N-terminal of the heavy chain (translocating domain) is inserted into the membrane of the synaptic vesicle, and the LC is subsequently translocated into the cytoplasm. 4) The activated LC selectively cleaves one of the SNARE proteins involved in ACh exocytosis, preventing the complete assembly of the synaptic fusion complex. BoNT/A cleaves SNAP-25, and BoNT/B cleaves the VAMP protein. Eventually, the toxin blocks the release of ACh into the synaptic space.
BoNT binds to specific receptors on the neuronal presynaptic surface and is internalized through receptor-mediated endocytosis. The acidic environment within the endocytic vesicles induces structural changes in BoNT, and the translocation of the BoNT’s catalytic domain, the light chain, into the cytosol takes place when the heavy chain inserts into the membrane of the synaptic vesicle. The light chain remains inactive and does not function while still connected to the rest of the toxin, but after translocation, the light chain is released from the rest of the toxin through the action of cleaving enzymes, such as heat shock protein 90.44,47 After internalization into the neuronal cytosol, the light chain of BoNT targets and cleaves one of three SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) proteins necessary for vesicular trafficking and neurotransmitter release: VAMP, SNAP25, and syntaxin. This enzymatic activity enables BoNTs to exert their toxic effects. 48 BoNT serotypes A and E specifically cleave SNAP-25, while serotype C cleaves both syntaxin and SNAP-25. Serotypes B, D, F, and G of BoNT hydrolyze VAMP. As a result, these proteins become inactive due to BoNT action, leading to the prevention of acetylcholine release and causing reversible chemical paralysis of the muscles.41,44,47
The mechanisms of pain-relieving effect of botulinum neurotoxin
BoNT’s pain-relieving effects are caused by several major mechanisms other than its direct effect on reducing muscle contraction. One significant mechanism through which BoNT alleviates pain, particularly in neuropathic pain conditions, is by inhibiting the release of neurotransmitters involved in the pain pathway, including CGRP,49,50 glutamate,51,52 substance P,53,54 and ATP, 55 through the suppression of SNARE proteins. Moreover, BoNT inhibits the release of pain mediators from peripheral nerve terminals, dorsal root ganglia, and spinal cord neurons, thereby reducing inflammation.56,57 BoNT has been found to suppress glial cell activity, enhances microglial M2 phenotype, and reduces the release of pro-nociceptive and pro-inflammatory mediators. 58 BoNT also modulates the communication between glial cells and neurons, inhibits c-Fos expression, and modulates receptors and ion channels involved in pain signaling.59,60 Additionally, BoNT’s central anti-nociceptive effect is attributed to its ability to cleave SNAP-25 in various central regions, such as the brainstem, 61 motor neurons of the spinal cord,62,63 and superior colliculus. 64 This occurs because BoNT can be retrogradely transported from the peripheral injection site to the central region, allowing it to exert its effects on both peripheral and central neural levels.
The effect of botulinum neurotoxin on pain-related receptors
Summary of studies on BoNT effects on pain-related receptors.
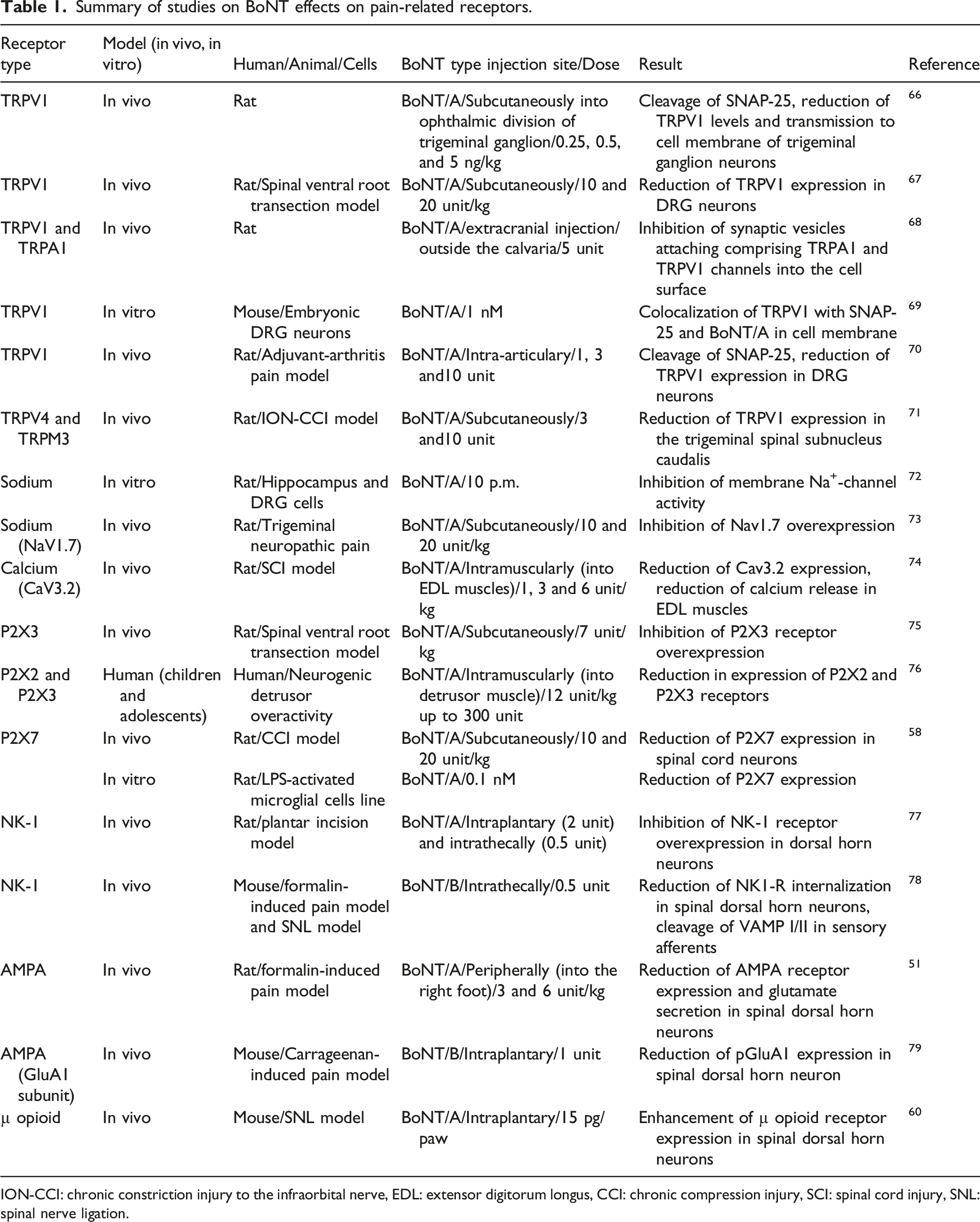
ION-CCI: chronic constriction injury to the infraorbital nerve, EDL: extensor digitorum longus, CCI: chronic compression injury, SCI: spinal cord injury, SNL: spinal nerve ligation.
Transient receptor potential ion channel
The transient receptor potential (TRP) ion channels, particularly TRPV1 and TRPA1, play a crucial role in various neuropathic pain conditions. 80 These non-selective ion channels are involved in hypersensitivity to chemical, mechanical, and thermal stimuli associated with inflammation and neuropathy. TRPV1 and TRPA1 are expressed together on nociceptive primary afferent fibers and, when activated by endogenous metabolites and natural mediators, they increase sensitivity to pain stimuli, contributing to the experience of pain.81,82 TRPV1 sensitization, a crucial mechanism in pain signaling, involves phosphorylation by various protein kinases (Ca2+/calmodulin-dependent protein kinase, PKA, PKC, PKG, and tyrosine kinase). Additionally, mediators like bradykinin, PGE2, ATP, and nerve growth factor can sensitize the TRPV1 channel. Another important aspect of pain signaling is the upregulation of TRPV1 expression, which occurs in pathological conditions such as peripheral nerve injuries, leading to increased TRP channel expression at the injury site.83–88
Studies have demonstrated that BoNT/A can decrease TRPV1 expression in peripheral tissues. For example, Apostolidis et al. found that local administration of BoNT/A reduced TRPV1 expression in the human bladder. 89 Moreover, the activation of PKC can induce the translocation of TRPV1 channels to the cell membrane through SNARE-dependent exocytosis in cultured DRG neurons, and BoNT/A inhibits this translocation of TRPV1 to the neuronal surface. 90 BoNT/A appears to hinder the trafficking of TRPV1 to cell membranes by cleaving the SNARE protein complex, leading to a decrease in TRPV1 expression.
BoNT/A can prevent the translocation of TRPV1 to cell membranes through two potential mechanisms. Firstly, it inhibits the phosphorylation of TRPV1, particularly at the Y200 site. 91 It has been observed that blocking SNAP25 activity reduces the delivery of NGF-induced TRPV1 to the cell membrane. 92 BoNT/A inhibits TRPV1 phosphorylation by cleaving SNAP25, which can lead to a decrease in TRPV1 levels. Additionally, BoNT/A affects TRPV1 expression through changes in cell membrane trafficking. In a study by Shimizu et al., subcutaneous injection of BoNT/A in rats’ ophthalmic branch of the trigeminal ganglion area resulted in SNAP25 cleavage and suppressed TRPV1 transmission to the plasma membrane in trigeminal ganglion neurons. RT-PCR analysis showed that BoNT/A did not reduce TRPV1 expression at the transcriptional level. These findings indicate that BoNT/A attenuates TRPV1 expression by inhibiting its cell membrane trafficking. 66 Similarly, Lizu Xiao and colleagues evaluated the effects of BoNT/A on TRPV1 expression in the DRG of a ventral root transaction-evoked neuropathic pain model in rats. They showed that TRPV1 protein levels were significantly upregulated in DRG neurons and that subcutaneous injection of BoNT/A abolished TRPV1 overexpression and attenuated hyperalgesia. 67
BoNT/A’s prophylactic efficacy in chronic migraine depends on reducing the sensitivity of meningeal nociceptors to TRPA1 and TRPV1 channel stimulation. Injecting BoNT/A outside the calvaria in rats successfully suppressed the responses of meningeal C-fiber nociceptors when capsaicin, a TRPV1 agonist, and mustard oil, a TRPA1 agonist, were used to stimulate intracranial dural receptive fields. Extracranial application of BoNT/A reduces the expression of TRPV1 and TRPA1 receptors in dural nerve endings. Administering BoNT/A along suture lines is more effective than muscle administration in inhibiting the activation of meningeal nociceptors through TRPV1 and TRPA1 channels. These findings suggest that injecting BoNT/A near the extracranial nerve endings of meningeal nociceptors can alleviate sensitivity by preventing the attachment of synaptic vesicles containing TRPA1 and TRPV1 channels to the plasma membrane of collateral branches of the same axon. 68
The interaction between the TRPV1 channel and BoNT/A is a critical factor for the analgesic effect of BoNT/A. Functional and structural interactions between the BoNT/A and TRPV1 receptors were observed in the cultured mouse DRG neurons. Following BoNT/A exposure, TRPV1 channels colocalized with cleaved SNAP-25 and BoNT/A in the plasma membrane of neurons. Moreover, TRPV1 bocking using a specific antibody reduced BoNT/A-induced SNAP-25 cleavage. These results showed that BoNT/A interacts structurally with TRPV1 receptors and that this interaction could functionally change the BoNT⁄A activity and TRPV1 levels. 69 In the adjuvant-arthritis pain model of rats, a study found a correlation between the analgesic effect of BoNT/A and TRPV1 levels. Intra-articular injection of BoNT/A dose-dependently reversed nociceptive behaviors, cleaved SNAP-25, and decreased TRPV1 expression in DRG neurons. The analgesic effect of BoNT/A was supported by the co-localization of cleaved SNAP-25 with TRPV1 channels and TRPV1 channels with CGRP in DRG neurons. Additionally, a single intra-articular injection of BoNT/A cleaved SNAP-25 in ipsilateral DRG neurons, indicating retrograde axonal transport. These findings suggest that BoNT/A may act as an anti-nociceptive agent by reducing TRPV1 expression through the inhibition of its cell membrane trafficking. 70 In a rat model of trigeminal neuralgia, Yi Zhang et al. discovered that the analgesic effect of BoNT/A might be linked to the reduction of TRPV4 and TRPM3 levels. In this model, TRPV4 and TRPM3 expression was increased in the trigeminal spinal subnucleus caudalis. The co-overexpression of TRPV4 and TRPM3 can lead to mechanical hyperalgesia. However, subcutaneous injection of BoNT/A reversed hyperalgesia by suppressing the overexpression of TRPV4 and TRPM3 in the central nervous system. 71 These findings suggest that the analgesic effect of BoNT/A involves the inhibition of TRP channel levels, although the exact mechanism remains unclear.
Sodium channel
Voltage-gated sodium channels (NaVs) are important receptors involved in transmitting and amplifying pain signals. 93 The nervous system has nine different isoforms of sodium channels (NaV1.1 to NaV1.9) with distinct properties and distribution.73,94 Sodium channels are implicated in various peripheral neuropathies. 95 Specifically, NaV1.3, NaV1.7, NaV1.8, and NaV1.9 have a critical role in activating nociceptors. 96 Sodium channels regulate the initial depolarization and propagation of action potentials, so changes in their expression and function impact neuronal excitability. 97 Nerve injury can modify sodium channel properties and expression, and alterations in sodium currents significantly contribute to the hyperexcitability observed in neuropathic pain conditions. 98
The deactivation of sodium channels appears to be one of the mechanisms by which BoNT/A can modulate pain behavior. Min-Chul Shin et al. demonstrated that BoNT/A blocked sodium channel activity in central and peripheral neuronal cells, reducing sodium current in cultured hippocampal and DRG cells. 72 Furthermore, subcutaneous injection of BoNT/A significantly reduced mechanical allodynia in rats with trigeminal neuropathic pain. Additionally, BoNT/A effectively inhibited the overexpression of Nav1.7 in the trigeminal ganglion of these nerve-injured animals. These findings suggest that the anti-nociceptive effects of BoNT/A involve suppressing NaV1.7 expression in the trigeminal ganglion. 73
Calcium channel
Voltage-gated calcium channels are important in the pain pathway, activating the action potential and triggering many physiological cytoplasmic processes. 99 Neurons express different classes of calcium channels, which are categorized into low and high-voltage-activated channels based on the activation voltage. 100 The low voltage-activated channels, also known as T-types or CaV3, contribute to neuronal excitability in the descending and ascending pain pathways within the brain, spinal dorsal horn, and primary afferent neurons.99,101–103 T-type calcium channels are enhanced in afferent pain fibers in several chronic pain disorders, such as diabetic neuropathy,104,105 traumatic nerve injury,106–108 and toxic neuropathies induced by chemotherapy.109–112 The CaV3.2 isoform is the most notable among the calcium channels involved in the pain process, and studies confirm it as a target for treating pain conditions. 99 The CaV3.2 channel is highly expressed in DRG neurons, and its contribution to chronic pain pathophysiology has been demonstrated in many reports.113,114 The current and excitability of T-type calcium channels were found to be enhanced in DRG cells of neuropathic pain models.115,116 Studies have revealed that suppression of the T-type calcium channel in spinal dorsal horn neurons might mediated analgesia in rat neuropathic pain models.99,117 These findings suggest the role of the T-type calcium channel in pain processing mediation.
BoNTs may help treat neuropathic pain by inhibiting the T-type calcium channel. Kening Ma et al., showed that BoNT/A reduced the expression of CaV3.2, leading to improved muscle spasticity in a rat SCI model. After SCI, calcium levels were elevated in the extensor digitorum longus (EDL) muscles. Injection of BoNT/A into EDL muscles alleviated symptoms, reduced CaV3.2 expression, and attenuated calcium release in a dose-dependent manner. These data suggest that BoNT/A might prevent SCI-evoked muscle spasticity by acting on the CaV3.2 calcium channel. 74
Purinergic receptor
The ATP signaling pathway is implicated in pain signal processing, with stimulation of ATP purinergic receptors in spinal cord neurons increases pain behavior in animal model. 118 Extracellular ATP activates two superfamilies of surface P2 purinergic receptors: ionotropic receptor channels (P2XRs) and the metabotropic G protein-coupled receptors (P2YRs), which are divided into seven and eight subtypes, respectively. These receptors are present in various cell types and play a vital role in regulating neurotransmission. 119 Several studies have shown a correlation between P2Y expression levels and neuropathic pain conditions, as nerve injuries can lead to dysregulation of P2Y receptor expression, resulting in neuropathic pain. 120 P2X channels, found in nerve terminals of peripheral tissues and the central terminal of DRG and trigeminal ganglia, are implicate in the generation and maintenance of pain. The involvement of P2X channels in pain has been demonstrated in several studies.119,121 Certain P2X receptor subtypes, including P2X2, P2X3, P2X4, and P2X7, play various roles in the pathophysiology of pain.121,122 For example, P2X3 receptors are critical in chronic inflammatory and neuropathic pain. 123 In a mouse model of chronic muscle hyperalgesia, a selective P2X3 receptor antagonist blocked P2X3 receptors and inhibited acute muscle hyperalgesia. 124
Studies have shown that BoNT can alleviate neuropathic pain syndrome by affecting P2X receptors. BoNT/A has been found to reduce hyperalgesia in a rat neuropathic pain model by inhibiting the overexpression of P2X3 receptors in DRG neurons. 75 In another study, BoNT/A reduced the expression levels of muscular P2X2 and P2X3 receptors in adolescents and children with neurogenic detrusor activity. 76 Additionally, BoNT/A has been shown to induce microglial M2 polarization and increase pain threshold by reducing spinal levels of P2X7 receptor in a rat model of neuropathic pain caused by chronic compression injury. Furthermore, in an in vitro inflammatory model, BoNT/A elevated microglial M2 polarization in LPS-activated microglial cells by inhibiting P2X7 receptor levels. 58
Neurokinin-1 receptor
The NK-1 receptors are activated by the tachykinin family of peptides, with the highest affinity to substance P. They are involved in neuropathic pain conditions and are present in peripheral tissues and the central nervous system.125,126 Stimulation of NK-1 receptors maintains hypersensitivity to pain,127,128 and in rodent neuropathic pain models, NK-1 receptor antagonists have been shown to block substance P-evoked activation of spinal cord neurons and inhibit pain transmission. 125
BoNT/A inhibits neuropathic pain by affecting the NK1 receptor internalization in the pain nerve terminal. In a rat plantar incision model, intraplantar and intrathecal administration of BoNT/A alleviated mechanical pain hypersensitivity and inhibited the increase of NK1 receptors in dorsal horn neurons. 77 BoNT has also been found to decrease collagen secretion by suppressing the NK1 receptor signaling pathway in fibroblasts activated during scar formation in cutaneous neurogenic inflammation. 129 In a mouse model of formalin-induced neuropathic pain, intrathecal injection of BoNT/B alleviated nociceptive behavior by reducing NK1-R internalization and the cleavage of VAMP I/II in sensory afferents. Additionally, in mouse models of neuropathic pain, deletion of genes encoding the NK1 receptor inhibited the analgesic function of BoNT/A. 78
Glutamate receptor
Glutamate function is mediated by two types of receptors: ionotropic and metabotropic glutamate receptors. The ionotropic glutamate receptors (iGluRs) family, including AMPA, NMDA, and kainite receptor, is responsible for fast excitatory neurotransmission. In contrast, the metabotropic glutamate receptors (mGluRs) family comprises eight subtypes, and is implicated in slow modulating synaptic transmission.130,131 The role of iGluRs in transmitting nociceptive stimuli has been well established. Blocking iGluRs through AMPA receptor, kainite receptor, and NMDA receptor antagonists has been shown to reduce nociceptive transmission in various experimental pain models. 132 Spinal AMPA receptors play a crucial role in neuropathic pain pathogenesis,51,133–137 and AMPA receptor antagonists have been found to reduce pain behavior in animal models of neuropathic pain.51,138 Clinically used AMPA receptor inhibitors have been effective in treating humans with chronic neuropathic pain. 139
BoNT/A along with AMPA receptor antagonists, has been shown to reduce the expression of AMPA receptors in spinal dorsal horn neurons. In a study by Bin Hong and colleagues, peripheral injection of BoNT/A decreased the function and expression of AMPA receptors in dorsal horn neurons through retrograde transportation to the spinal cord. BoNT/A alleviated formalin-induced nociceptive hypersensitivity in rats and reduced glutamate release from dorsal horn neurons by cleavage of SNAP-25. They suggested that peripheral administration of BoNT/A alters the expression of AMPA receptors and glutamate secretion from spinal cord dorsal horn neurons, which might be responsible for its central anti-nociceptive functions. 51 AMPA receptors are composed of four subunits, GluA1-GluA4, each contributing specific properties to the receptor. In a mice model of inflammatory pain, BoNT/B was found to reduce pGluA1 expression, decrease carrageenan-induced allodynia, and prevent NMDA-induced phosphorylation of GluA1. This suggested that peripheral application of BoNT/B transported to the central region and inhibited the phosphorylation of AMPA receptor subunits. 79
NMDA receptor antagonists have been shown to alleviate symptoms of neuropathic pain in both experimental models and patients. 131 In a zebrafish orofacial neuropathic pain model, BoNT/A was shown to decrease nociceptive behavior and interact with the NMDA receptor. The analgesic function of BoNT/A was prevented by the NMDA receptor antagonist ketamine, and molecular data showed an interaction between BoNT/A and NMDA receptors, indicating the involvement of NMDA receptor mechanisms in the anti-nociceptive effects of BoNT/A in neuropathic pain conditions. 140
BoNT/A has been shown to alleviate neuropathic pain by modulating the expression and translocation of various receptors, including TRP ion channels, sodium channels, calcium channels, purinergic receptors, NK-1 receptor, and glutamate receptors. However, it is unclear whether these effects represent the primary mechanism by which BoNT/A exerts its analgesic effects. Further research is needed to fully understand the mechanisms by which BoNT/A reduces pain and to determine the relative contributions of these various mechanisms. Moreover, it is important to note that pain is a complex phenomenon that involves multiple physiological and psychological factors, and targeting a single receptor or pathway may not always be sufficient to effectively manage pain in all patients. A comprehensive approach that includes a combination of pharmacological and non-pharmacological interventions may be necessary to effectively manage neuropathic pain.
The role of opioidergic system in anti-nociceptive effect of botulinum neurotoxin
The opioid system plays an important role in pain experience and management, with opioid receptors widely distributed in peripheral and central nervous areas. The density of opioid receptors and the function of opioid peptides can change under different neuropathic pain conditions. 141 Studies have demonstrated that the availability of opioid receptors decreases in the brains of patients with chronic pain conditions,142–148 including reduced µ opioid receptor expression in cortical regions implicated in pain conditions.142,149,150 Additionally, downregulation of µ opioid receptor expression has been observed in cortical regions of a rat model of peripheral neuropathic pain. 151
The involvement of the endogenous opioid system in the central anti-nociceptive effect of BoNT/A in an inflammatory pain model has been demonstrated. In both sciatic nerve transaction and formalin-induced pain models, the peripheral anti-nociceptive function of BoNT/A was abolished by systemic (subcutaneously) and central (intrathecally) administration of naltrexone, a non-selective opioid antagonist. Additionally, the analgesic effect of BoNT/A was found to be prevented by intrathecal injection of naloxonazine, a selective µ receptor antagonist, indicating that the analgesic effect of BoNT/A is mediated by the µ opioid receptor. BoNT/A also downregulated c-Fos expression levels in spinal dorsal horn neurons, and this effect was inhibited by naltrexone. These findings suggest that the central analgesic effect of BoNT/A may be associated with endogenous opioid system activity. 152 The interaction between BoNT/A and the opioidergic system was also demonstrated in another study. In a mouse model of neuropathic pain, Intraplantar administration of BoNT/A inhibited morphine-evoked tolerance allodynia and increased the analgesic action of morphine in rats with sciatic nerve lesion-induced neuropathic pain. The expression of the µ opioid receptor was downregulated in dorsal horn neurons of mice with neuropathic pain under chronic morphine treatment, and BoNT/A restored the expression levels of the µ opioid receptor. These results indicate that the beneficial effect of BoNT/A in morphine-evoked tolerance is related to an increase in µ opioid receptor expression in neurons. 60
Association between the anti-nociceptive action of botulinum neurotoxin and GABAergic system
Gamma-aminobutyric acid (GABA) is the main inhibitory neurotransmitter in the brain and spinal cord dorsal horn. 153 GABAergic synaptic inhibition plays a critical inhibitory role in the transmission of nociceptive information in neuropathic pain.154–156 The inhibitory effect of GABA is mediated through its GABA-A ionotropic receptor and GABA-B metabotropic receptor. Several studies have demonstrated that GABAergic pathway plasticity is implicated in the generation and development of neuropathic pain after nerve injury, with GABA expression and neuronal activity being reduced in animal models of nerve injury. 157 The GABA-A receptor agonist has been found to abolish neuropathic pain behavior, indicating the critical role of GABAergic inhibitory pathways in modulating chronic pain conditions. 158
A study has demonstrated a correlation between the analgesic effect of BoNT/A and the GABA-A receptor. The systemic administration of bicuculline, a GABA-A receptor antagonist, was found to prevent the analgesic effect of peripheral BoNT/A injection in rat models of inflammatory and neuropathic pain. Additionally, bicuculline abolished the inhibitory effect of BoNT/A on c-Fos expression levels in spinal dorsal horn neurons. These findings suggest that the GABA-A receptor antagonist abolishes the anti-nociceptive activity of BoNT/A by reducing the GABAergic inhibition pathway in the dorsal horn of the spinal cord, which is essential in the initiation and maintenance of chronic pain. BoNT/A can reduce inhibitory GABA activity by cleaving SNAP-25 and suppressing neurotransmitter release. 159
Overall, the studies have shown that BoNT/A may modulate the endogenous opioid system and GABAergic pathways to reduce pain in animal models of neuropathic pain and in vitro experiments. It is also important to note that the opioid system and GABAergic pathways have complex interactions with other neurotransmitter systems and pathways involved in pain modulation.160–164 Therefore, the effects of BoNTs on these systems may be influenced by the activity of other systems and pathways, which may vary depending on the type and severity of the pain and individual patient factors. While the studies suggest a potential role for BoNTs in modulating the opioid system and GABAergic pathways to alleviate neuropathic pain, further research is needed to fully understand the mechanisms by which they work. Additional research is needed to better comprehend the complex interactions between different neurotransmitter systems and pathways involved in pain modulation and to develop more effective and comprehensive approaches to pain management.
Conclusions
BoNT has been employed as a therapeutic option for pain conditions, and its ability to alleviate pain is attributed to various significant mechanisms. One crucial mechanism involves the inhibition of pain-related receptors, which is associated with the inhibition of SNARE proteins. BoNT has the capacity to regulate the expression of TRP ion channels, sodium channels, calcium channels, purinergic receptors, NK-1 receptors, and glutamate receptors, which potentially contribute to its analgesic effects. Additionally, BoNT can modulate the endogenous opioid system and GABAergic pathways, thereby reducing pain sensation.
Nevertheless, although BoNT holds significant potential as a therapeutic strategy for neuropathic pain further investigation is necessary to gain a comprehensive understanding of the exact mechanism underlying the analgesic effects of BoNT. Furthermore, most of the studies performed on animal models, which may not precisely mirror human physiology. Consequently, the findings may not directly apply to humans. Additionally, certain studies are performed in vitro, potentially failing to fully capture the complexity of pain conditions in humans. As a result, the external validity of these studies and the generalizability of their findings may be limited.
Footnotes
Acknowledgments
We thank the Pain Research Center, Neuroscience Institute, Imam Khomeini Hospital Complex, Tehran University of Medical Sciences, Tehran, Iran
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.