
Abstract
Ca2+ imaging is frequently used in the investigation of sensory neuronal function and nociception. In vitro imaging of acutely dissociated sensory neurons using membrane-permeant fluorescent Ca2+ indicators remains the most common approach to study Ca2+ signalling in sensory neurons. Fluo4 is a popular choice of single-wavelength indicator due to its brightness, high affinity for Ca2+ and ease of use. However, unlike ratiometric indicators, the emission intensity from single-wavelength indicators can be affected by indicator concentration, optical path length, excitation intensity and detector efficiency. As such, without careful calibration, it can be difficult to draw inferences from differences in the magnitude of Ca2+ transients recorded using Fluo4. Here, we show that a method scarcely used in sensory neurophysiology – first proposed by Maravall and colleagues (2000) – can provide reliable estimates of absolute cytosolic Ca2+ concentration ([Ca2+]cyt) in acutely dissociated sensory neurons using Fluo4. This method is straightforward to implement; is applicable to any high-affinity single-wavelength Ca2+ indicator with a large dynamic range; and provides estimates of [Ca2+]cyt in line with other methods, including ratiometric imaging. Use of this method will improve the granularity of sensory neuron Ca2+ imaging data obtained with Fluo4.
Findings
Background
Measuring changes in [Ca2+]cyt is a commonly used method for determining the sensitivity of sensory neurons to different stimuli and investigating pro-nociceptive signalling pathways, with Fluo4 being a popular choice of single-wavelength Ca2+ indicator. As has been discussed in detail previously,
1
one can estimate [Ca2+]cyt from the optical properties of a fluorescent Ca2+ indicator using the following equation
2


It is apparent that if R is large, as is the case for Fluo4 (R ≈ 85-100
1
), then 1/R will become negligible (as 1/R << F/Fmax), and equation (2) can be rewritten as

The estimate of [Ca2+]cyt provided by equation (3) does not require the experimenter to find Fmin. Fmax can be found easily at the end of each experiment by applying 0.1% Triton-X in a bathing solution containing 10 mM Ca2+. As R and KD are properties intrinsic to the indicator, it is not necessary for them to be estimated for each experiment, 1 though it is important to consider that these parameters are sensitive to the intracellular environment (e.g., pH, ionic composition). 5 Therefore, adding a straightforward step to the end of each Ca2+ imaging experiment can provide an estimate of [Ca2+]cyt using Fluo4.
Estimating changes in [Ca2+]cyt
Ionomycin is often used to calibrate Ca2+ signals measured in sensory neurons and while this calibration provides some useful information, it cannot be used to estimate absolute [Ca2+]cyt. This is because ionomycin cannot raise [Ca2+]cyt sufficiently to obtain a true Fmax for the indicator. This is apparent if 50 mM KCl and 5 µM ionomycin are sequentially applied to sensory neurons in a bath solution containing 2 mM Ca2+ (Figure 1, trace i). Ionomycin application cannot have resulted in saturation of Fluo4 because the magnitude of Ca2+ transients evoked by KCl (black traces) exceeded those evoked by ionomycin. Similar results were obtained after raising bath [Ca2+] to 20 mM during the application of ionomycin, though the response to ionomycin was greater under these conditions (Figure 1, trace ii). Calibration of Ca2+ transients using ionomycin. Example traces showing the change in Fluo4 fluorescence over baseline (ΔF) during the application of 50 mM KCl and 5 µM ionomycin. KCl was applied in the presence of 2 mM bath Ca2+ (black traces), and ionomycin was applied in the presence of either 2 mM (blue trace) or 20 mM (orange trace) bath Ca2+. In both cases, the peak fluorescence evoked by KCl application was greater than that evoked by ionomycin application, showing that ionomycin application cannot have resulted in saturation of the indicator.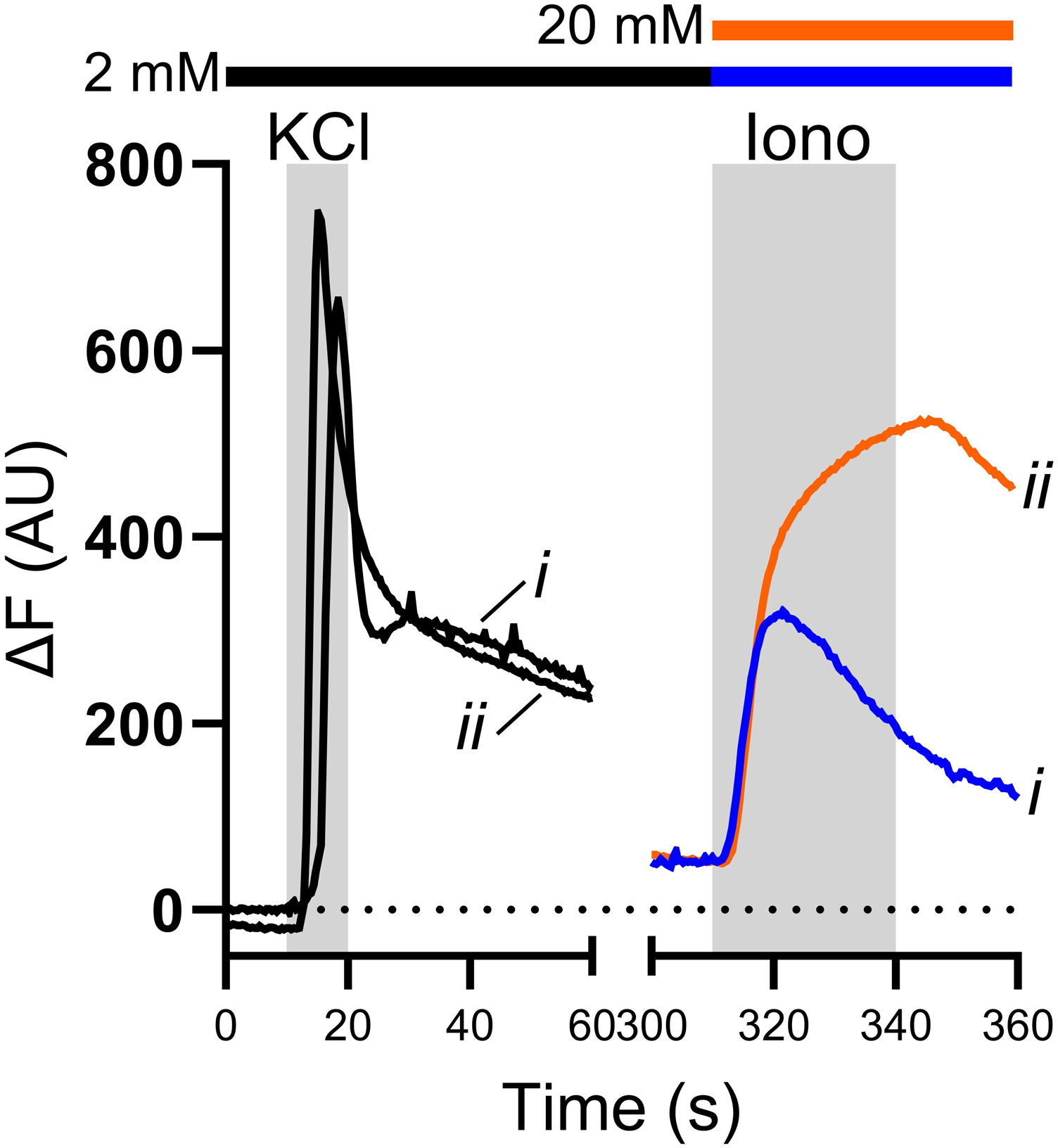
While KCl is useful for identifying viable neurons, its use as a calibration for the magnitude of Ca2+ signals in sensory neurons is limited because the magnitude of the response to KCl is dependent on voltage-gated Ca2+ channel function and cytosolic Ca2+ buffering, which may not be comparable between different sensory neuron subpopulations 6 or different experimental conditions.
The absolute Fmin or Fmax for Fluo4 – as with other indicators – can be found by applying 0.1% Triton-X (to lyse cells) with either a high concentration of a Ca2+ chelator (1 mM EGTA) in Ca2+-free bathing solution (Figure 2(a), grey trace), or 10 mM Ca2+-containing bathing solution (Figure 2(a), black trace), respectively. The transient increase in F following the application of Triton-X/EGTA in Ca2+-free bathing solution (Figure 2(a), grey trace) partly represents liberation of Ca2+ from intracellular stores. It is important to note that in the case of equation (3), as F tends towards Fmax, 1 – F/Fmax will tend towards zero and, hence, estimates of [Ca2+]cyt become unreasonably large. While F/Fmax < 90%, F/Fmax is approximately linear with [Ca2+]cyt and provides a reliable measure of [Ca2+]cyt (Figure 2(b)). Beyond 90%, F/Fmax no longer provides a reliable metric of [Ca2+]cyt and estimates become unreasonably large – as such, this has been dubbed the “zone of unreliability” (Figure 2(b), red shaded region
7
). It is, therefore, important to ensure that F measured during an experiment remains within the approximately linear region of equation (3) and does not exceed 90% F/Fmax. Measuring changes in [Ca2+]cyt in sensory neurons using Fluo4. (a) Example traces from two neurons showing the application of 0.1% Triton-X to sensory neurons in the presence of either 10 mM bath Ca2+ (black trace) or 0 mM bath Ca2+ and 1 mM EGTA (grey trace). The peak of the black trace shows Fmax for this neuron, while the minimum of the grey trace shows Fmin. The peak of the grey trace shows the liberation of Ca2+ from intracellular stores as the neuron is lysed. (b) The relationship between F/Fmax and [Ca2+]/KD. For F/Fmax < 90%, [Ca2+]/KD remains small and approximately linear (inset). However, as F/Fmax becomes larger (>90%), estimates of [Ca2+] become very large and unreliable (red shaded region). (c) Example trace showing uncorrected Fluo4 fluorescence from a single neuron during the application of 50 mM KCl (2 mM bath Ca2+) and 0.1% Triton-X (10 mM bath Ca2+). (d) Example traces showing Fluo4 fluorescence normalised to Fmax for five randomly-selected neurons during the application of 50 mM KCl. Traces in (d)-(f) are colour-coded to show data from the same individual neurons. Inset: grouped data showing peak F/Fmax for all neurons imaged (n = 33 neurons from three independent DRG preparations, dashed line shows F/Fmax = 0.9). (e) Example traces showing [Ca2+]cyt for five randomly-selected neurons during the application of 50 mM KCl. [Ca2+]cyt was calculated from equation (1) using Fmin = 5.852 AU (from the experiment in (a)). (f) Example traces showing [Ca2+]cyt for five randomly-selected neurons during the application of 50 mM KCl. [Ca2+]cyt was calculated from equation (3). (g) Scatterplot comparing estimates for [Ca2+]cyt calculated using equations (1) and (3) for the five neurons in (d)-(f). Each dataset was fit with a straight line (R2 > 0.99); the average slope was 1.006 ± 0.0004 and each line passed approximately through the origin, showing a high congruence in the estimates of [Ca2+]cyt provided by equations (1) and (3). (h) Heatmap showing the percentage error in the estimate of [Ca2+]cyt between equations (1) and (3) for each neuron (black bar shows KCl application). (i) Grouped data showing the average baseline [Ca2+]cyt calculated using equations (1) and (3) for each neuron (n = 33 neurons from three independent DRG preparations). Data analysed using a Mann-Whitney U-test. (j) Grouped data showing the peak [Ca2+]cyt calculated using equations (1) and (3) for each neuron (n = 33 neurons from three independent DRG preparations). Data analysed using a Mann-Whitney U-test. (k) Example traces showing [Ca2+]cyt (calculated using equation (3)) during the application of 250 nM BK (n = 13 neurons from two independent DRG preparations). (L) Grouped data showing average basal (pre-BK; mean of first 10 s of recording) and peak (post-BK) [Ca2+]cyt (n = 13 neurons from two independent DRG preparations).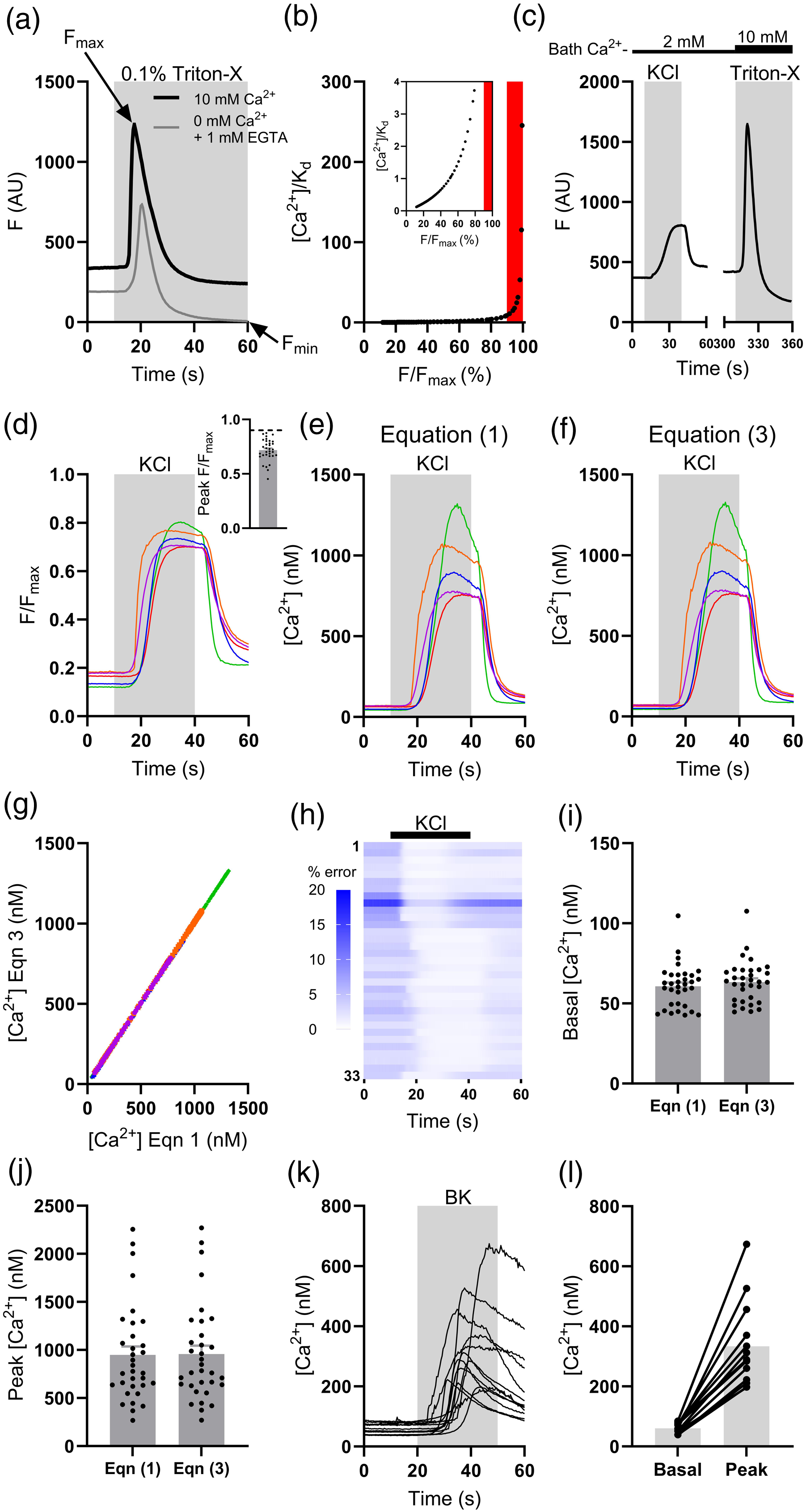
To test the utility of equation (3) for measuring [Ca2+]cyt in sensory neurons, 50 mM KCl was used to induce depolarisation-dependent Ca2+ transients (n = 33 neurons, 2 mM bath Ca2+, Figure 2(c)). Estimates of [Ca2+]cyt calculated using equations (1) and (3) were compared; the value for Fmin used in equation (1) was found in the experiment shown in Figure 2(a). Fmax was found for each neuron at the end of each experiment. The peak F evoked by KCl application remained below 90% of that evoked by Triton-X/10 mM Ca2+; peak F/Fmax was 0.72 ± 0.02 (range: 0.45-0.87, Figure 2(d)). As such, reliable estimates of [Ca2+]cyt were possible. Figure 2(e) and (f) show example traces of [Ca2+]cyt obtained using equations (1) and (3), respectively. The data look qualitatively very similar. The congruence in the estimates of [Ca2+]cyt found using equations (1) and (3) (Figure 2(g)) demonstrates that the dynamic range of Fluo4 is of sufficient magnitude to make its reciprocal negligibly small relative to F/Fmax. The percentage error in the estimates of [Ca2+]cyt provided by equations (1) and (3) was lowest during the peak of the KCl-evoked Ca2+ transient (0.97 ± 0.06%) and highest during baseline (4.36 ± 0.43%, Figure 2(h)). No difference in the estimated average baseline [Ca2+]cyt was found between equations (1) and (3) (p = .26, Figure 2(i)). The estimates of peak [Ca2+]cyt provided by equations (1) and (3) were also no different (p = .78, Figure 2(j)). Importantly, estimates calculated from equation (3) of both baseline (63.4 ± 2.3 nM) and peak (956.9 ± 88.1 nM) [Ca2+]cyt agreed with previous estimates made using ratiometric Ca2+ imaging.8–11 Changes in [Ca2+]cyt evoked by 50 mM KCl are relatively large, but this method can also be used to calibrate smaller changes in [Ca2+]cyt. The application of the algogenic mediator bradykinin (BK, 250 nM, 2 mM bath Ca2+) to sensory neurons raised [Ca2+]cyt – calculated using equation (3) – from 60.3 ± 4.8 nM to 333.3 ± 39.6 nM (n = 13 neurons, Figure 2(k) and (l)).
Basal [Ca2+]cyt
Multiple factors, including ageing
11
and inflammation,
12
can affect basal [Ca2+]cyt in rodent sensory neurons. We have examined the effect of soma size on resting [Ca2+]cyt in sensory neurons (Figure 3(a); soma area distribution shown in inset). Neurons were parsed into subgroups by soma area (A): small (A <400 µm2, n = 86), medium (400 < A <1000 µm2, n = 129) and large (A >1000 µm2, n = 78). Although there was no difference in resting [Ca2+]cyt between small- and medium-area neurons (73.0 ± 3.8 nM vs 74.3 ± 3.8 nM, p > .99, Figure 3(b)), basal [Ca2+]cyt in large-area neurons (52.8 ± 2.6 nM) was lower than both small- (p = .0002) and medium-area (p < .0001) neurons (Figure 3(b)). Average basal [Ca2+]cyt across all neurons in this sample was 68.2 ± 2.2 nM (n = 293 neurons from three independent DRG preparations), in agreement with previous estimates in rodent sensory neurons.8–14 A modestly reduced resting [Ca2+]cyt in large-diameter sensory neurons has been observed in rat,
6
but corroborating observations in mouse sensory neurons are lacking. It is not immediately clear why large-area sensory neurons would exhibit a lower resting [Ca2+]cyt, though it could be due to more efficient [Ca2+] buffering. To test this possibility, another sample of neurons (n = 112 neurons from three independent DRG preparations) was stimulated with 50 mM KCl and the decay of evoked Ca2+ transients (expressed as the time constant for the decay of the transient) was analysed across different neuronal sizes. There was a modest negative correlation between soma area and time constant (r = −0.26, p = .0061, Figure 3(c)). KCl-evoked Ca2+ transients in small-area neurons exhibited a time constant of 13.0 ± 1.2 s (n = 39), compared to 8.0 ± 1.0 s (n = 23) in large-area neurons (p = .0068, Figure 3(d)). KCl-evoked Ca2+ transients in medium-area neurons exhibited an intermediate time constant (10.4 ± 0.8 s, n = 50) which was indistinguishable from that in small- (p = .25) and large-area (p = .26) neurons (Figure 3(d)). The more rapid decay of KCl-evoked Ca2+ transients in large-area neurons (compared with small-area neurons) is consistent with a greater capacity for Ca2+ buffering.
6
Measuring basal [Ca2+]cyt in sensory neurons parsed by soma size. (a) Scatterplot showing soma area and average basal [Ca2+]cyt (n = 293 neurons from three independent DRG preparations). Inset: frequency distribution of soma sizes. (b) Grouped data showing the average basal [Ca2+]cyt for small, medium and large sensory neuronal soma. Data analysed using a Kruskal-Wallis ANOVA with Dunn’s post-hoc tests. (c) Scatterplot showing soma area versus time constant for the decay of KCl-evoked Ca2+ transients (n = 112 neurons from three independent DRG preparations). Line of best fit shown to illustrate modest negative correlation (solid black line with 95% confidence limits shown as dashed lines). Inset: traces showing the decay of KCl-evoked Ca2+ transients in exemplar small- (orange), medium- (blue), and large-area (purple) neurons. (d) Grouped data showing the time constant for KCl-evoked Ca2+ transients in small, medium and large sensory neuronal soma. Data analysed using a Kruskal-Wallis ANOVA with Dunn’s post-hoc tests.
In summary, the method put forward by Maravall and colleagues 1 provides a straightforward, easy-to-implement and – importantly – reliable protocol for estimating [Ca2+]cyt in sensory neurons using a high dynamic range single-wavelength indicator without the need to estimate Fmin. Estimates of [Ca2+]cyt in sensory neurons made using this method with Fluo4 are well in-line with estimates made using other methods, including ratiometric Ca2+ imaging. Fmin need not be estimated because the dynamic range of Fluo4 is sufficiently large, as evidenced by the close alignment of estimates of [Ca2+]cyt between equations (1) and (3). Use of this method broadens the utility of Fluo4 and will enable more rigorous quantification of Ca2+ signalling in sensory neurons using Fluo4.
Methods
Preparation of sensory neurons
All animal work was carried out in accordance with the Animals (Scientific Procedures) Act 1986. Mice (C57Bl/6, Charles River) were housed in groups of up to six littermates under a 12 h light/dark cycle with bedding material, enrichment (e.g., igloos, tunnels, etc) and ad libitum access to food and water. All mice used were male aged 8-14 weeks.
Sensory neurons were prepared as described previously.15–17 Briefly, dorsal root ganglia (DRG, T12-L5) were removed from the spinal column and enzymatically digested in collagenase (1 mg/mL) and trypsin (1 mg/mL). DRG were then mechanically dispersed by trituration through a pipette tip. Dispersed neurons were seeded onto glass-bottomed culture dishes coated with poly-
Ca2+ imaging
Growth medium was aspirated from culture dishes and neurons were incubated with 10 µM Fluo4-AM for 30-45 min at room temperature (shielded from light). Neurons were then washed and bathed in extracellular bath solution containing (in mM): 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 4
All imaging was carried out at room temperature. Images were acquired at 2.5 fps with 100 ms exposure using a Retiga Electro CCD camera (QImaging, BC, Canada). Fluo4 was excited by a 470 nm light source (Cairn Research, UK) and emission at 520 nm was recorded using µManager. 18 For experiments using 50 mM KCl as a stimulus, KCl was superfused for 30 s following a 10 s baseline. For experiments using 250 nM bradykinin as a stimulus, bradykinin was superfused for 30 s following a 20 s baseline. At the end of all experiments, 0.1% Triton-X in 10 mM Ca2+ bath solution was superfused to lyse cells and yield an estimate for Fmax for each neuron. Fmin was estimated in one experiment by applying 0.1% Triton-X to neurons bathed in Ca2+-free bath solution containing 1 mM EGTA.
Data analysis
Regions of interest were manually traced around neurons and the average pixel intensity per region per frame was calculated using ImageJ. After the subtraction of background fluorescence, measured fluorescence values for each neuron were calibrated to [Ca2+] using equations (1) and (3) using a KD for Fluo4 of 325 nM.
Datasets were scrutinised to ensure that they met the assumptions of parametric analyses (normality tested using the Shapiro-Wilk test; equality of variances tested using F-tests) and, where appropriate, non-parametric, rank-based alternatives were used. Details of statistical tests used are in the figure legends. To find the time constant for the decay of KCl-evoked Ca2+ transients, data were fit with a one-phase exponential decay. The time constant gives the time for the transient to decay by a factor of 1/e, that is, ∼37% of peak. This is equivalent to T50/ln(2), where T50 is the half-life of the decay of the transient, that is, the time for the transient to decay by a factor of 1/2. Analysis was carried out in GraphPad Prism (GraphPad Inc.). Grouped data are displayed as mean ± standard error.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Biotechnology and Biological Sciences Research Council (BB/M01194/1).