
Abstract
Tumor metastasis to bone is often accompanied by a severe pain syndrome (cancer-induced bone pain, CIBP) that is frequently unresponsive to analgesics, which markedly reduces patient quality of life and cancer treatment tolerance in patients. Prolonged pain can induce hypersensitivity via spinal plasticity, and several recent studies have implicated the involvement of vascular endothelial growth factor-A (VEGF-A) signaling in this process. Here, we speculated that CIBP is associated with VEGF-A/VEGFR2 signaling in the spinal cord. A mouse model of CIBP was established by intramedullary injection of Lewis lung carcinoma (LLC) cells in the mouse femur. Pain sensitization and potential amelioration via VEGF-A/VEGFR2 blockade were measured using paw withdrawal threshold to mechanical stimulation and paw withdrawal latency to thermal. Spinal VEGF-A/VEGFR2 signaling was blocked by intrathecal injection of the VEGF-A antibody or the specific VEGFR2 inhibitor ZM323881. Changes in the expression levels of VEGF-A, VEGFR2, and other pain-related signaling factors were measured using western blotting and immunofluorescence staining. Mice after LLC injection demonstrated mechanical allodynia and thermal hyperalgesia, both of which were suppressed via anti-VEGF-A antibody or ZM323881. Conversely, the intrathecal injection of exogenous VEGF-A was sufficient to cause pain hypersensitivity in naïve mice via the VEGFR2-mediated activation of protein kinase C. Moreover, the spinal blockade of VEGF-A or VEGFR2 also suppressed N-methyl-D-aspartate receptor (NMDAR) activation and downstream Ca2+-dependent signaling. Thus, spinal VEGF-A/VEGFR2/NMDAR signaling pathways may be critical mediators of CIBP.
Keywords
Introduction
Many advanced tumors, including breast, lung, kidney, and prostate tumors, are prone to bone metastasis, thereby frequently resulting in a severe pain syndrome termed cancer-induced bone pain (CIPB).1 Indeed, approximately 75% of patients with advanced-stage cancer suffer from moderate to severe CIBP, which further diminishes the quality of life and may even reduce anti-cancer treatment tolerance and survival rates.2,3 Furthermore, anti-tumor therapies such as radiotherapy and chemotherapy often fail to reduce CIBP. 4 Therefore, further studies of the underlying cellular and molecular mechanisms are required to identify feasible treatment targets for CIBP.
Vascular endothelial growth factor-A (VEGF-A) is a member of the VEGF family of multifunction growth factors that also includes VEGF-B, VEGF-C, VEGF-D, VEGF-E, and placental growth factor (PIGF). The most extensively studied physiological response to VEGF-A is angiogenesis mediated by binding to VEGF receptor 2 (VEGFR2), as VEGF-A/VEGFR2-induced angiogenesis is critical for the growth and survival of tumors.5,6 Furthermore, although the VEGF-A/VEGFR2 signaling is implicated in chronic pain development, the underlying cellular, and molecular mechanisms are still unclear. N-methyl-D-aspartate (NMDA) receptors (NMDARs) and protein kinase C (PKC) also contribute to the induction and maintenance of central pain sensitization associated with CIBP. 7 Based on these findings, we hypothesized that CIPB is mediated by enhanced VEGF-A/VEGFR2 signaling and ensuing hyperactivation of PKC and NMDARs in the spinal cord.
In the present study, we first examined the expression levels of VEGF-A and VEGFR2 in the spinal dorsal horn are increased in CIBP model mice and associated with behavioral manifestations of pain sensitization (mechanical allodynia and thermal hyperalgesia). We then examined the pharmacologic inhibition of VEGF-A/VEGFR2 signaling can ameliorate pain sensitization and NMDAR activation. Taken together these results can identify new potential therapeutic targets for CIBP management.
Materials and methods
Cell culture
The murine Lewis lung carcinoma (LLC) cell line was purchased from the Shanghai Cell Bank, Chinese Academy of Sciences. The cells were seeded in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 25 mM glucose and 10% fetal bovine serum (FBS; Gibco). Cultures were maintained in a humidified incubator (Thermo, Waltham, MA, USA) at 37°C under a 5% CO2/95% O2 atmosphere and passaged at 1:2 every 1–2 days.
Experimental animals
Specific-pathogen-free (SPF) male C57BL/6 mice weighing 20–25 g were obtained from the Experimental Animal Center, Jinan, China, and housed five per cage or fewer at ∼22 ± 0.5°C under a 12-h/12-h light/dark cycle with ad libitum access to food and water. Animals were acclimated to these conditions for 3 days before the initiation of experiments.
All experimental protocols were approved by the Animal Protection and Use Committee of Xuzhou Medical University and complied with the National Institute of Health Guide for the Care and Use of Laboratory Animals and the International Association for the Study of Pain guidelines for pain research. 8
Mouse model of CIBP
LLC cells were detached using 0.125% trypsin, collected by centrifugation, and resuspended at 2×108/μL in sterile phosphate-buffered saline (PBS). The cells were then injected in the mouse according to a previously reported method with modifications. 9 In brief, mice were anesthetized with 2% sodium pentobarbital (50 mg/kg) by intraperitoneal (i.p.) injection in the supine position. The superficial skin of the left leg was shaved and disinfected with 75% (v/v) ethanol, and 10 μL of LLC cell suspension was slowly injected into the medullary cavity of the left distal femur using a 20-μL microinjection syringe. The syringe tip was left in place for 90 s after injection to allow cells to fill the bone cavity. After removing the needle, the injection hole was sealed with sterile bone wax to prevent cell leakage. Sham control mice were injected with 10 μL sterile PBS into the left femur.
Drug administration
Mouse VEGF-A neutralizing antibody (AF493) and control antibody (normal goat immunoglobulin G, AB-108-C) were purchased from R&D Systems (Minneapolis, MN, USA), and the VEGFR2 inhibitor ZM323881 (S2896) was purchased from Selleck Chemicals (Shanghai, China). Mouse recombinant VEGF-A (CYT336) was purchased from ProSpec (East Brunswick, NJ, USA). ZM323881 was first dissolved in dimethyl sulfoxide and then diluted in sterile PBS, whereas all other drugs were dissolved directly in sterile PBS. All drugs (10 μL each) were injected into the cerebral spinal fluid via lumbar puncture with doses determined in preliminary experiments. 10 Intrathecal injection was performed using a 10-μL Hamilton syringe with a 30-gauge needle in the L5-6 interspace. The successful insertion of the needle into the lumbar subarachnoid space was confirmed by a brisk tail and/or paw flick. Reagent doses and treatment time points are specified in the individual figure legends.
Pain behavioral tests
Assessment of mechanical allodynia
The paw withdrawal threshold (PWT) was measured as an index of mechanical pain tolerance using von Frey filaments (Aesthesio ®, Danmic Global, San Jose, CA, USA). 11 All tests were performed in a polymethyl methacrylate box with a wire net floor. In brief, mice were first habituated to the experimental environment for three consecutive days and then again for 20 min immediately before testing. A series of filaments (0.02, 0.04, 0.07, 0.4, 0.6, 1.0, and 1.4 g) was applied to the mid-plantar surface of the hind paw with pressure sufficient to bend each for 3–4 s or induce a paw to withdrawal reflex, flinch, or lick within 4 s. The 50% PWT was calculated using Dixon’s up and down method. 12
Assessment of thermal hyperalgesia
Paw withdrawal latency (PWL) in response to radiant heat stimulation was measured as an index of thermal pain tolerance.13,14 The tests were conducted in a polymethyl methacrylate box placed on the glass plate of Plantar Analgesia Meter (IITC Life Science, Woodland Hills, CA, USA) equipped with a controlled radiant heat source. Animals were exposed to the apparatus for three consecutive days and again for 20 min before each test. The radiant heat was terminated when the mouse withdrew its hind paw. A 20-s cutoff was set to avoid tissue damage. The pain threshold of the hind paw was first measured for each mouse. This same heat stimulus was then repeated three times to each hind paw with a 10-min interval.
Bone histology
Sham control and CIBP model mice were sacrificed on day 14 post-injection via decapitation under anesthesia with 2% pentobarbital (150 mg/kg, i.p.). The bilateral hind femurs were excised, fixed in 4% paraformaldehyde overnight at 4°C, demineralization in 10% ethylenediamine tetraacetic acid for 2 weeks, embedded in paraffin, and cut into 4-mm thick sections using a rotary microtome. Sections were subsequently stained with hematoxylin and eosin (HE) and examined under a light microscope to assess tumor cell infiltration and bone tissue destruction.
Western blotting
The L4–L6 spinal cord segments were quickly removed from deeply anesthetized mice after laminectomy and stored under liquid nitrogen until further processing. Each spinal cord tissue sample was homogenized in pre-cooled radioimmunoprecipitation assay lysis buffer (Beyotime, Shanghai, China) containing a protease inhibitor cocktail and phosphatase inhibitor. The homogenates were centrifuged at 12,000 r/min for 15 min at 4°C and the supernatants were collected for the assessment of total protein concentration using a bicinchoninic acid protein assay kit (Thermo Scientific). The equal amount of lysate protein per gel lane was then separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto 0.22-μm polyvinylidene difluoride membranes (Millipore, Burlington, MA, USA). Membranes were blocked in Tris-buffered saline (TBS) containing 5% nonfat dry milk and 0.1% Tween-20 for 1 h at room temperature and then incubated overnight at 4°C with the indicated primary antibodies. Membranes were washed three times (5 min/wash) in TBS plus Tween-20 (TBST) and then incubated with specific horseradish peroxidase conjugated goat anti-rabbit secondary antibody (1:10,000, Proteintech, Wuhan, China). The following rabbit primary antibodies were used for western blotting (all at 1:1,000 unless otherwise indicated): anti-phospho-p44/42 mitogen-activated protein kinase (MAPK) (ERK1/2) (Thr202/Thr204; Cell Signaling Technology, Danvers, MA, USA), anti-phospho-CaMKII (Thr286; Cell Signaling Technology), anti-phospho-cyclic-AMP response element-bingding (CREB)CREB (Ser133; Cell Signaling Technology), anti-phospho-NR2B (Tyr1472; Cell Signaling Technology), anti-VEGF-A (Abcam, Cambridge, UK), anti-VEGFR2 (Cell Signaling Technology), anti-VEGFR2 (phospho-Y951, Abcam), and anti-GAPDH (1:10,000, Proteintech) as the gel loading control. The intensities of target protein bands were quantified using ImageJ (NIH, Bethesda, MD, USA) and normalized to the GAPDH band intensity. Fold-changes are expressed relative to the corresponding control group.
Immunofluorescence and image analysis
Mice were deeply anesthetized with pentobarbital sodium (150 mg/kg, i.p.) and perfused transcardially with PBS followed by 4% paraformaldehyde. The L4-6 spinal cord segments were excised, post-fixed in 4% paraformaldehyde overnight at 4°C, preserved in 0.1 M phosphate buffer containing 30% sucrose at 4°C, and then cut into 20-μm transverse sections using a cryostat. Sections were blocked in TBS containing 10% normal donkey serum for 2 h at room temperature, incubated with the indicated primary antibodies overnight at 4°C, and washedthree times in TBS (5 min/wash). The antibodies used for immunostaining were as follows: mouse VEGF164 antibody (1:200, AF-493-SP, R&D Systems), rabbit anti-VEGFR2 (1:500, sc-505, Santa Cruz), mouse anti-neuronal nuclear (NeuN) (1:1000, MAB377, Millipore), rabbit anti-GFAP (1:200, A14673, ABclonal), rabbit anti-IBA-1 (1:200, 48,934s, Cell Signaling Technology), mouse anti-GFAP (1:10,00, #3670, Cell Signaling Technology), goat anti-IBA1 (1:500, Novus, St Charles, MI, USA), mouse anti-OLIG2 (1:500, MABN50, Millipore), goat anti-CGRP (1:500, ab36001, Abcam), anti-isolectin GS-IB4 (1:500, Invitrogen, Waltham, MA, USA), anti-MAP2 (1:200, MAB3418, Sigma, Millipore), and mouse anti-CD31 (1:200, AF3628, R&D Systems). The corresponding secondary antibodies were added to the sections for 1 h at 20°C–25°C. Immunolabeled sections were mounted on slides for observation under a confocal scanning laser microscope (FV1000; Olympus, Tokyo, Japan). The mean fluorescence intensity of VEGFR2 and numbers of VEGFR2-immunopositive neurons were determined in 16 spinal cord sections from four mice using ImageJ.
Statistical analyses
All data are expressed as means ± standard deviation. GraphPad Prism 8 (GraphPad Software, La Jolla, CA, USA) was used to conductfor all statistical analyses. Group differences in western blot and immunofluorescence measurements were evaluated using one-way analysis of variance (ANOVA), followed by post hoc Dunnett’s multiple comparisons tests. Group differences in pain metrics were evaluated using two-way ANOVA.
Results
Intrafemoral inoculation of LLC cells induces bone destruction and pain hypersensitivity
The injection of LLC cells into the mouse femoral cavity was used to establish a model of CIBP. The cortical and trabecular bone tissues of the sham mouse had normal cells and clear boundaries in the HE staining (Figure 1 (a)). The HE of sections derived from excised femur 14 days post-inoculation revealed that tumor cells were still packed in the bone marrow cavity with progressive invasion into the surrounding bone and the partial destruction of cortical and trabecular bone tissues (Figure 1 (b)). By contrast, these pathological characteristics were absent in the contralateral femur and the bilateral femurs of the sham control mice injected with saline (Figure 1 (a)). The inoculation of LLC cells into the femur also induced mechanical allodynia and thermal hyperalgesia of the ipsilateral hind paw but not the contralateral hind paw of tumor-bearing mice or the bilateral hind paws of sham control mice, which is consistent with previous studies.15,16 PWT in response to von Frey hair stimulation was significantly reduced on day 3 post-inoculation and remained at a low level from days 4–14 (the day of sacrifice for histopathology) (Figure 1 (c)). Likewise, PWL in response to radiant heat stimulation was significantly reduced on day 5 post-inoculation and maintained until day 14 post-inoculation (Figure 1 (d)). In addition, no changes in PWT and PWL were detected in the contralateral hind paw of model mice or bilateral hind paws of sham control mice. Intrafemoral injection of Lewis lung carcinoma (LLC) cells in mice led to the mimicking of the histopathology of bone metastasis and induced pain hypersensitivity characteristic of cancer-induced bone pain (CIBP). (A and B) Hematoxylin and eosin (HE) staining showed that the injected (ipsilateral) distal femur was infiltrated and severely damaged by LLC cells on day 14. (C and D) Reduced paw withdrawal threshold (PWT) to mechanical stimulation (C) and paw withdrawal latency (PWL) to thermal stimulation (D) in the ipsilateral hind paw following LLC cell injection, which is consistent with the induction of mechanical allodynia and thermal hyperalgesia, respectively. Results in (C) and (D) are expressed as mean (±SD) of six mice per group. *p < 0.05, **p< 0.01 and ***p < 0.001 versus sham control mice. Scale bars: 20 μm.
Upregulation of VEGF-A and VEGFR2 in the spinal cord following LLC inoculation
It is now widely recognized that the functions of VEGFs are not limited to angiogenesis and regulation of vascular permeability, but in fact may be relevant to the development of cancer pain.17,18 Furthermore, changes in VEGF signaling have been reported in a plethora of diseases in association with pain development, including cancer,10,16,19 rheumatoid arthritis,
20
and diabetic complications.21,22 Likewise, western blotting revealed upregulated expression of both VEGF-A and VEGFR2 proteins in the spinal cord from days 5–14 post-inoculation, compared with control mice (Figure 2 (a) and 2 (b)). Tumor cell injection increased the expression of vascular endothelial growth factor-A (VEGF-A) and vascular endothelial growth factor receptor 2 (VEGFR2) in the spinal dorsal horn. (A and B) Western blots showing the time course of VEGF-A and VEGFR2 upregulation in the spinal cord after intrafemoral LLC cell injection. (A) Representative bands. (B) Densitometric results. Results in (B) are expressed as the mean (±SD) of four mice per group. *p < 0.05, **p < 0.01 and ***p < 0.001 versus sham mice. Expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the gel loading control. Blockade of VEGF-A/VEGFR2 signaling attenuates mechanical allodynia and thermal hyperalgesia in CIBP model mice.
To examine whether the increased spinal expression levels of VEGF-A and VEGFR2 following LLC cell inoculation are associated with behavioral pain sensitization, we tested the effects of a VEGF-A neutralizing antibody and a VEGFR2 inhibitor. A single intrathecal injection of either anti-VEGF-A (2 μg in 10 μL) or ZM323881 (10 μL of 10 nM) on day 12 post-inoculation attenuated both mechanical allodynia (Figure (3) and 3 (b)) and thermal hyperalgesia (Figure 3 (c) and 3(d)). In addition, the daily intrathecal injection of anti-VEGF-A or ZM323881 from post-inoculation days 12–14 significantly delayed the progression of mechanical allodynia (Figure 3 (e) and 3(f)) and thermal hyperalgesia (Figure 3 (g) and 3 (h)). By contrast, there were no significant differences in PWT and PWL between the sham controls receiving anti-VEGF-A or a control IgG and between sham controls receiving ZM323881 or vehicle (DMSO). Besides, following three successive injections of VEGF-A antibody or VEGFR2 inhibitor, the analgesic effect could last mostly 6 h and disappear by 24 hours (Figure 3 (i) and 3 (l)). Intrathecal (i.t.) injection of VEGF-A antibody (anti-VEGF-A) or VEGFR2 inhibitor (ZM323881) attenuated pain sensitization following LLC cell injection. (A–D) A single administration of anti-VEGF-A (0.2 μg in 10 μL i.t.) or ZM323881 (10 μL of 10 nM, i.t.) on day 12 post-injection significantly delayed or attenuated mechanical allodynia (A, C) and thermal hyperalgesia (B, D) compared to corresponding controls (IgG for anti-VEGF-A and 1% DMSO for ZM323881). Results are expressed as mean (±SD) of six mice per group. ***p < 0.001 versus sham + IgG (or DMSO), #p < 0.05, ##p < 0.01, ###p < 0.001 versus tumor + IgG (or DMSO). (E to H) Daily administration of anti-VEGF-A or ZM323881 (same doses as in A–D) from day 12–14 post-injection significantly attenuated mechanical allodynia (E, F) and hyperalgesia (G, H). Behavioral tests were performed 4 h after each injection. Results are presented as the mean (±SD) of six mice per group. *p < 0.05, ***p < 0.001 versus sham + IgG (or DMSO); #p < 0.05, ##p < 0.01, ###p < 0.001 versus tumor + IgG (or DMSO). (I to L) The third administration of anti-VEGF-A (0.2 μg in 10 μL i.t.) or ZM323881 (10 μL of 10 nM, i.t.) on day 14 post-injection significantly delayed or attenuated mechanical allodynia (I, K) and thermal hyperalgesia (J, L) compared to corresponding controls (IgG for anti-VEGF-A and 1% DMSO for ZM323881). The analgesia effect could last nearly 6 h and disappear within 24 h. Results are presented as the mean (±SD) of six mice per group. ***p < 0.001 versus sham + IgG (or DMSO); #p < 0.05, ##p < 0.01, ###p < 0.001 versus tumor + IgG (or DMSO).
These results strongly suggest that VEGF-A/VEGFR2 signaling directly contributes to pain sensitization during the progression of experimental bone cancer in mice.
Intrathecal injection of exogenous VEGF-A elicits pain hypersensitivity in naïve mice
To further provide supports for the contribution of enhanced spinal VEGF-A/VEGFR2 signaling to pain sensitization, we examined the effects of recombinant VEGF-A in naïve mice. The intrathecal injection of VEGF-A reduced the PWT (Figure 4 (a)) and PWL (Figure 4 (b) within 2 h, and this pain hypersensitivity lasted for at least 12 h before returning to baseline. Moreover, both acute mechanical allodynia and thermal hyperalgesia were prevented by intrathecal injection of ZM323881 30 min before VEGF-A injection (Figure 4(a) and 4 (b)). These results are consistent with a previous report that VEGF-A injection triggers VEGFR2-dependent mechanical and thermal hyperalgesia in naïve rats,
10
and further confirms that enhanced spinal VEGF-A/VEGFR2 signaling contributes to pain sensitization in CIBP model mice. Exogenous VEGF induces VEGFR2-dependent pain sensitization in naïve mice. (A, B) Intrathecal injection of exogenous recombinant VEGF-A (0.1 μg) induced time-dependent mechanical allodynia (A) and thermal hyperalgesia (B) in naïve mice, while pre-administration of the VEGFR2 inhibitor ZM323881 (10 nmol) 30 min before VEGF-A injection partially attenuated these effects. Results are the average (±SD) of six mice per group. **p < 0.01, ***p < 0.001 versus phosphate-buffered saline (PBS) injection group; #p < 0.05, ##p < 0.01, and ###p < 0.001 versus VEGF-A injection group.
VEGF-A and VEGFR2 are co-expressed in spinal neurons after LLC inoculation
The implantation of tumor cells within the femur or tibia has been reported to induce sustained stimulation of primary afferent fibers projecting to the L4-6 spinal cord, resulting in central pain sensitization.23, 24 Likewise, the results presented in Figures 1 and 2 demonstrate that VEGF-A/VEGFR2 signaling contributes to CIBP in a mouse model. To identify the potential anatomical substrates for this association between VEGF-A/VEGFR2 signaling and pain sensitization, we first examined cell type-specific expression of VEGF-A and VEGFR2 via double-staining immunofluorescence VEGF-A immunoreactivity predominantly colocalized with NeuN (Figure 5(a)), occasionally colocalized with GFAP (Figure 5(b)) and IBA-1 (Figure 5(c)). Consistent with previous studies,10, 25 VEGFR2 colocalized predominantly with NeuN (Figure 6(a)), but not with markers of microtubule-associated protein 2 (MAP2) for neuronal dendrites (Figure 6 (b)), calcitonin gene-related peptide (CGRP) for peptidergic primary afferent terminals, isolectin b4 (Ib4) for non-peptidergic primary afferent terminals (Figure 6 (c)), platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31) for vascular endothelia (Figure 6 (d)), oligodendrocyte lineage transcription factor 2 (OLIG2) for oligodendrocytes (Figure 6 (e)), glial fibrillary acidic protein (GFAP) for astrocytes (Figure 6 (f)), and ionized calcium-binding adaptor molecule-1 (IBA-1) for microglia (Figure 6(g)). Further, in contrast to previous findings in CIBP model rats,10 VEGFR2 did not colocalize with the microglial marker ionized calcium-binding adaptor molecule-1 (Figure 6(g)). Given that VEGF-A and VEGFR2 were both expressed in spinal neurons, we next speculated that VEGF-A/VEGFR2 may mediate neuron sensitization in CIBP conditions. Localization of VEGF-A to the cell bodies of the spinal dorsal horn. (A to C) Dual immunofluorescence staining for VEGF-A and a cell-specific marker in the spinal dorsal horn. The cellular markers used were NeuN for neuronal nuclei (NeuN), glial fibrillary acidic protein (GFAP) for astrocytes, and ionized calcium-binding adaptor molecule 1 (IBA-1) for microglia. Tissue sections were prepared 14 days after intrafemoral LLC cell injection. VEGF-A immunoreactivity predominantly colocalized with NeuN (A) and occasionally colocalized with GFAP (B) and IBA-1 (C). Scale bars: 20 μm. Localization of VEGFR2 to the cell bodies of the spinal dorsal horn. (A–G) Dual immunofluorescence staining for VEGFR2 and a cell-specific marker in the spinal dorsal horn. The cellular markers used were NeuN for neuronal nuclei (NeuN), microtubule-associated protein 2 (MAP2) for neuronal dendrites (B), calcitonin gene-related peptide (CGRP) for peptidergic primary afferent terminals, isolectin b4 (Ib4) for non-peptidergic primary afferent terminals (C), platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31) for vascular endothelia (D), oligodendrocyte lineage transcription factor 2 (OLIG2) for oligodendrocytes (E), glial fibrillary acidic protein (GFAP) for astrocytes (F), and ionized calcium-binding adaptor molecule 1 (IBA-1) for microglia (G). Tissue sections were prepared 14 days after intrafemoral LLC cell injection. VEGFR2 immunoreactivity was mainly colocalized with the neuronal nucleus marker NeuN (A). Scale bars: 20 μm.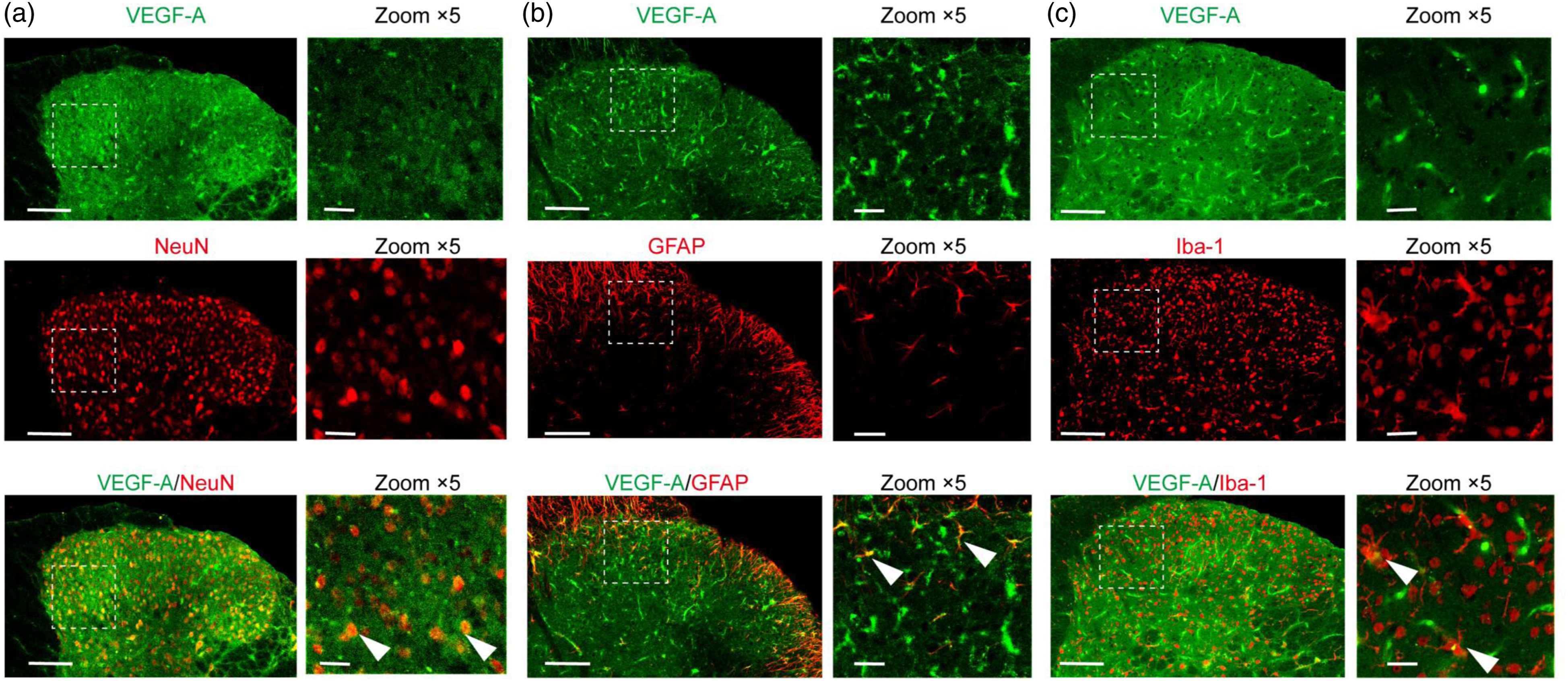

VEGF-A/VEGFR2/PKC signaling activates NMDA receptors and downstream calcium-dependent molecular cascades in CIBP model mice
We then explored the molecular pathways underlying VEGF-A/VEGFR2-evoked pain sensitization. Six major intracellular signaling pathways are known to be activated by VEGFR2: phospholipase Cγ (PLCγ), protein kinase C (PKC), phosphoinositide 3-kinase (PI3K), mitogen-activated protein kinase (MAPK), focal adhesion kinase (FAK), and Src family kinase.26-28 In the present study, we focused on VEGF-A/VEGFR2/PKC signaling as previous immunofluorescence staining confirmed that PKC was demonstrated predominantly expressed in neurons
10
which was overlapped with VEGFR2 (Figure (6)). Further, we examined the contributions of spinal NMDARs owing to strong evidence implicating upregulated NMDAR phosphorylation and NMDAR--mediated plasticity in central sensitization.
29
For instance, the spinal central sensitization and continuous pain are associated with the upregulated phosphorylation of NMDAR subunit NR1 (p-NR1).30,31 In turn, NMDAR activation leads to enhanced postsynaptic Ca2+ influx and downstream signal transduction via extracellular signal-regulated kinase (ERK1/2), calcium/calmodulin-dependent protein kinase II (CaMKII), and cyclic-AMP response element-binding protein (CREB). Protein kinase C can enhance NR1 expression in spinal neurons
32
and also promote NMDA receptor activity indirectly by increasing AMPA receptor activity via ERK1/2 and CaMKII-mediated autophosphorylation.33,34 In addition, CREB drives NMDAR subunit overexpression in chronic pain.
35
Therefore, we speculated that VEGF-A/VEGFR2/PKC signaling-dependent NMDAR hyperactivation and the ensuing CAMKII/CREB-driven plasticity mediates pain sensitization in the CIBP model mice. Consistent with this notion, the intrathecal injection of anti-VEGF-A or ZM323881 significantly blunted the expression levels of phosphorylated NMDA receptor subunit 2B (p-NR2B) (Figure 7 (a)), p-ERK (Figure 7 (b)), p-CaMKII (Figure 7 (c)), and p-CREB (Figure 7 (d)). Intrathecal injection of anti-VEGF-A or VEGFR2 inhibitor following LLC cell injection suppressed the activation of NMDA receptors and downstream signaling molecules in the spinal cord. Intrathecal anti-VEGF-A or ZM323881injection significantly attenuated the LLC cell injection-induced upregulation of phosphorylated (p) NMDA receptor subunit 2B (p-NR2B) (A), extracellular signal-regulated kinase (p-ERK1/2) (B), calcium/calmodulin-dependent protein kinase II (p-CAMKII) (C), and cyclic-AMP response element-binding protein (p-CREB) (D) in the spinal dorsal horn. (A to D) VEGF-A neutralizing antibody (Ab; 2 μg, intrathecal) or ZM323881 (ZM, 10 nM, intrathecal) was injected once daily on days 12, 13, and 14 post-tumor inoculations. The spinal tissues were obtained 1 h after the last injection on day 14. Results are expressed as mean ± SD of spinal cord sections from four mice per group. ***p < 0.001 versus sham group; #p < 0.05, ##p < 0.01, and ###p < 0.001 versus LLC cell injection group.
Discussion
Patients with metastatic breast, lung, or prostate cancer frequently develop CIBP that manifests as a combination of background and breakthrough pain. 36 Tumor and related stromal cells release factors that sensitize and activate bone nociceptors, injure sensory nerve fibers, induce the release of growth factors, and contribute to peripheral pain sensitization. 1 Several recent studies have also shown that VEGF-A/VEGFR2 signaling triggers the sensitization of primary sensory neurons via activating the P2X2/3 receptors or calcineurin in the dorsal root ganglion.37-39 However, few studies examined whether VEGF-A/VEGFR2 signaling contributes to bone cancer pain and spinal sensitization in the CIBP model mice. Here we reported that the expression levels of VEGF-A and VEGFR2 in spinal dorsal horn were upregulated progressively following CIBP model induction (intrafemoral LLC cell injection), and intrathecal injection of anti-VEGF-A or a VEGFR2 inhibitor suppressed mechanical allodynia and thermal hyperalgesia in CIBP model mice. Moreover, intrathecal injection of VEGF-A induced pain hypersensitivity in naïve mice and this response was mitigated by co-treatment with a VEGFR2 inhibitor. Taken together, our results suggest that VEGF-A/VEGFR2 signaling contributes to the development and maintenance of CIBP.
In addition, we identified that VEGF-A was primarily expressed in the spinal neurons, occasionally in spinal astrocytes and microglia, while VEGFR2 was primarily expressed in spinal neurons. A previous study localized VEGFR2 to both the central terminals of primary sensory neurons and microglia in tumor-bearing mice, 10 while we found that VEGFR2 was primarily expressed by spinal neurons. This discrepancy may be explained by distinct levels of neuroinflammation in these two models as microglial VEGFR2 expression is strongly linked to chronic inflammatory pain. Most of the studies using experimental models of cancer-induced pain, inflammatory pain, and neuropathic pain addressed that the anti-nociceptive role derived from the inhibition of VEGF-A/VEGFR2, but not VEGFR1.10,15,25,8,40. In addition, VEGFR2 is the main receptor of VEGF-A and its activation plays a significant role in cell proliferation, migration, angiogenesis, and permeabilization, 26 and great success has been made to develop VEGF-A/VEGFR2 signaling inhibitors for cancer treatment.6,40,41 However, only one study reported that the anti-nociceptive role derived from the inhibition of VEGF-A/VEGFR2, but not VEGFR2 in neoplastic pain. 16 So we used the cancer-induced pain model to explore the role of VEGF-A/VEGFR2 which would inform the design and development of new pharmacological strategies.
PKC is a major activator of NMDARs and several studies have also demonstrated PKC upregulation after tumor cell inoculation.10, 42,43 It has been reported that PKC can not only indirectly activate NMDA receptors by promoting the synergistic action of AMPA receptors; it also phosphorylates NMDA receptors directly. 44,45 Given that protein kinase C is primarily expressed in neurons, 43 we hypothesized that VEGF-A/VEGFR2 enhances neuronal NMDA receptor function through the PKC signaling pathway. Therefore, we examined whether VEGF-A/VEGFR2/PKC signaling activates NMDARs and the downstream Ca2+-dependent signaling pathways. Indeed, western blot analysis demonstrated a substantial upregulation of p-NR2B, p-ERK1/2, p-CaKII, and p-CREB following LLC cell injection. Moreover, both the pain sensitization and activation of these signaling cascades were blocked by anti-VEGF-A antibody or a VEGFR2 inhibitor. These results suggest that CIBP results from the PKC-mediated transactivation of VEGF-A/VEGFR2 signaling and hyperactivation of NMDARs containing NR2B in peripheral and spinal neurons, which in turn lead to activation of downstream Ca2+-dependent signaling cascades underlying synaptoplastic changes that enhance pain transmission. Therefore, VEGF-A/VEGFR2 signaling may be an effective therapeutic target for CIBP.
This study has several limitations that must be addressed in follow-up studies. First, we did not directly measure NMDA currents or examine changes in excitatory postsynaptic current strength among peripheral sensory or spinal neurons. Research has shown that VEGF-A induces the nociceptive sensitization of peripheral sensory neurons in human patients with cancer and mouse cancer models via VEGFR1 rather than VEGFR2. 16 Moreover, while the majority of evidence supports the pro-nociceptive effects of VEGF-A, some studies have found an anti-nociceptive effect of a VEGF-A splice variant in neuropathic pain models.46,47 Thus, the mechanisms underlying VEGF-A alternative splicing and functions of VEGFR1 warrant further studies in CIBP.
Taken together, these findings suggest that vascular endothelial growth factor-A/vascular endothelial growth factor receptor 2 signaling may be an effective target not only for cancer treatment but also for CIBP.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Key Project of the Natural Science Foundation of Jiangsu Education Department (16KJA320002 to Wen Shen).