
Abstract
Chronic low back pain (cLBP) that cannot be attributable to a specific pathoanatomical change is associated with high personal and societal costs. Still, the underlying mechanism that causes and sustains such a phenotype is largely unknown. Emerging evidence suggests that epigenetic changes play a role in chronic pain conditions. Using reduced representation bisulfite sequencing (RRBS), we evaluated DNA methylation profiles of adults with non-specific cLBP (n = 50) and pain-free controls (n = 48). We identified 28,325 hypermethylated and 36,936 hypomethylated CpG sites (p < 0.05). After correcting for multiple testing, we identified 159 DMRs (q < 0.01and methylation difference > 10%), the majority of which were located in CpG island (50%) and promoter regions (48%) on the associated genes. The genes associated with the differentially methylated regions were highly enriched in biological processes that have previously been implicated in immune signaling, endochondral ossification, and G-protein coupled transmissions. Our findings support inflammatory alterations and the role of bone maturation in cLBP. This study suggests that epigenetic regulation has an important role in the pathophysiology of non-specific cLBP and a basis for future studies in biomarker development and targeted interventions.
Keywords
Globally, low back pain has an annual prevalence of about 38% (affects approximately 2.88 billion people) and is the leading cause of disability.1,2 Athough many people with low back pain do not seek care, it remains one of the most common reasons for visits to primary care providers. 3 For some individuals, the pain resolves within 4–6 weeks. However, for many individuals the low back pain progresses to chronic low back pain (cLBP), lasting more than 12 weeks. 2 Furthermore, for about 80 percent of patients with cLBP, the diagnosis cannot be attributed to a recognizable, specific pathoanatomical change such as trauma, osteoporosis, ankylosis spondylitis, and chronic inflammatory disease.3,4 As a result, these individuals are classified as having non-specific cLBP.3,4 Non-specific cLBP affects individuals of all race, sex, and socioeconomic status. It has been associated with mood disorders, decreased productivity, and reduced quality of life. At present, treatment and interventions for cLBP often fail to provide sufficient levels of pain relief, and full functional restoration is rarely achieved.2,3,5 Lack of human mechanistic studies that can offer treatment targets has been identified as a potential barrier to the development of effective interventions.6,7
Like most chronic health conditions, cLBP has both genetic and environmental influences.8–10 Twin studies estimate that about 46 to 75% of low back pain is heritable,9,11 and environmental factors account for approximately 50% of the variance in pain perception and prevalence.12–15 Environmental factors such as increased physical and psychological stress (e.g., physically demanding job, 16 low job control, 17 depression,18,19 anxiety, 8 catastrophizing,8,18 and perceived discrimination 20 ) have been associated with cLBP. Thus, a mechanism such as epigenetics that links stressful environmental factors to alterations in genetic expression may play a significant role in cLBP. 21
Emerging evidence attest to the role of epigenetics in many chronic pain conditions.15,22–26 Epigenetics represents a mechanism by which the environment can directly enhance or suppress gene expression without alterations of the primary DNA sequence.27,28 For instance, DNA methylation is a stable epigenetic change in which cytosine (C) nucleotides in CpG dinucleotides are methylated. Recent studies suggest that epigenetic mechanisms underlie the development of many chronic pain conditions such as fibromyalgia,15,22 chronic postoperative pain,23,24 and cLBP.25,26 Secreted protein acidic and cysteine-rich (SPARC) protein is required for the calcification of collagen in bone. Tajerian et al. examined the relationship between spinal disc degeneration, pain severity, SPARC gene expression, and DNA methylation of the SPARC promoter in mice and human with severe cLBP. They showed that, in vitro, increased DNA methylation of the SPARC promoter decreases the expression of the SPARC gene. In addition, DNA methylation of 5 out of 12 CpGs in the SPARC promoter correlated with cLBP intensity. 29 Understanding epigenetic differences between individuals with cLBP and individuals without cLBP can increase our knowledge of the pathogenesis and prognosis of cLBP.
Epigenome-wide reduced representation bisulfite sequencing (RRBS) provides robust study design, that can be exploited to aid in understanding subtle variations in cLBP pathogenesis. 30 However, the power of this approach has not yet been utilized to examine clinical cLBP. The aim of this study was to identify DNA methylation changes that are distinctive to adults with cLBP. We hypothesized that differential DNA methylation profiles could identify important pathways involved in cLBP.
Methods
Participants
Whole blood was collected from participants enrolled in an on-going study: Examining Racial And SocioEconomic Disparities in cLBP (ERASED) study (R01MD010441). Criteria for enrollment in the study are detailed in previously published work.31,32 Briefly, 50 adult males and females, ages 19 to 85 years, with non-specific cLBP, were chronologically enrolled in the study between June 2018 and September 2019. To be considered cLBP, the pain must have persisted for at least three consecutive months and resulted in pain on at least half the days in the past six months. Pain phenotype data was collected from participants with cLBP using self-report and confirmed diagnosis in the electronic medical record. Participants were identified as having non-specific cLBP based on joint clinical practice guidelines from the American Colleges of Physicians and the American Pain Society. 33 In addition, pain severity and interference were assessed using the Brief Pain Inventory (BPI) – Short Form, a widely used pain management tool. 34 Participants were excluded if they had a medical condition that could confound the interpretation of the phenotype or epigenomic changes, e.g., low back pain attributable to infection, trauma, malignancy, or ankylosing spondylitis, systemic infection, chronic inflammatory disease (e.g., rheumatoid arthritis, systemic lupus erythematosus, fibromyalgia), poorly controlled diabetes, neurological disorders (e.g., Parkinson’s, multiple sclerosis, and epilepsy) and any severe psychiatric disorder requiring hospitalization within the past 12 months.
Additionally, 48 pain-free controls (PFC) were recruited and enrolled in the study using the same procedure those with cLBP. Eligibility for the PFC group included men and women between 19–85 years of age who could read and write in English with no recent history of pain at any location and not pregnant or breastfeeding. The same exclusion criteria were applied to the PFC. The University of Alabama at Birmingham (UAB) Institutional Review Board for Human Subject Research approved all aspects of the study. All participants received informed consent information, had all questions answered, and provided written consent.
DNA extraction and RRBS
Venous blood samples were collected into ethylene-diamine-tetra-acetic acid (EDTA) tubes and centrifuged at 2500 x g for 10 mins at room temperature. Plasma and buffy coat were isolated, aliquoted, and stored at -80°C until DNA extraction. Genomic DNA was extracted from the buffy coat by the UAB Center for Clinical and Translational Sciences according to the Gentra Puregene DNA Purification Protocol (Qiagen, Valencia, CA, USA). Extracted DNA was quantified on the NanoDrop 2000, and purity was determined through spectrophotometry. All samples had a 260 nm to 280 nm ratio of >1.8.
RRBS libraries were prepared by the Heflin Center for Genomic Sciences at UAB using the Ovation RRBS Methyl-Seq kit (Tecan Genomics, Redwood City, CA, USA). High-quality genomic DNA was digested with MspI restriction enzymes. Fragments of the DNA were ligated with adaptors and C-to-T-converted (bisulfite conversion) strands sequenced on the NextSeq 500 platform from Illumina to generate raw RRBS reads. A Qubit fluorometer was used to assess the quantity and quality of the libraries.
DNA methylation and data pre-processing
The raw RRBS reads were cleaned, and adapters removed using Trim Galore. 35 The trimmed reads were then aligned and mapped to the human reference genome (hg19) using Bismark. 36 After discarding reads with multiple mapping, reads with unique alignments were identified. Following Bismark alignment and mapping, we used methylKit to identify the CpG sites, normalize, and perform differential methylation (hypo/hypermethylation). 37 In our sample, the mapping efficiency of Bismark aligning sequences to hg19 ranged from 71 to 75%, and the methylKit C methylated in the CpG context ranged from 38.6 to 58.7%.
Data analysis
The methylation level of each cytosine (C) was estimated by
Functional annotation and enrichment analysis
MethylKit was used to annotate differentially methylated bases, and regions to gene components based on the nearest transcriptional start site using the University of California, Santa Cruz Genome Brower (http://genome.ucsc.edu/; hg19). Features of a CpG island included CG length of at least 200 bp, CG fractions > 0.5, and an observed to expected CpG ratio of > 0.6. Adjacent positions to CpG islands of 2,000 bp were identified as CpG shores. Genomic features including promoters, exons, introns, and intergenic regions were identified using Genomic Features. 39
To examine the potential functions of the identified differentially methylated regions and annotated gene, functional enrichment analysis was performed using Gene Ontology (GO) pathway analyses.
Results
Sample characteristics
Our final sample included 50 adults with cLBP and 48 PFCs. The mean age of participants was 42.1 ± 13.8 years; half of them self-identified as non-Hispanic White (50.5%) and female (52.5%). There was no significant group differences in terms of mean age, race, or gender of particpants. As expected participants with a history of cLBP had a significantly higher pain severity score (4.6 ± 2.4) on the BPI compared to those without cLBP (p < 0.0001). Details of the demographic characteristics of participants are summarized in Table 1.
Characteristics of study participants.

Individual sample analysis
The Euclidean distance was calculated based on the individual sample methylation patterns. Wards minimum variance was used to hierarchically cluster the entire sample, as depicted in the dendrogram (Figure 1). The sample shows clustering, especially among individuals with cLBP (blue color).

Comparison of the variation in methylation patterns between individual samples using Ward’s methods. Notes: test/blue = cLBP and ctrl/red = PFC.
Also, we performed principal component analysis (PCA) to reduce a large number of data points into principal components (PCs). Our PCA provided two major PCs (PC1 and PC2), indicating two potential DNA methylation patterns. As depicted in Figure 2, the methylation patterns between control and test samples could be similar because of the overlap between the red cluster (control samples) and the blue cluster (test samples). Interestingly, several other test samples do not overlap with the control samples (the non-overlapping data points), suggesting that there may be different DNA methylation patterns in test samples compared to the control samples.
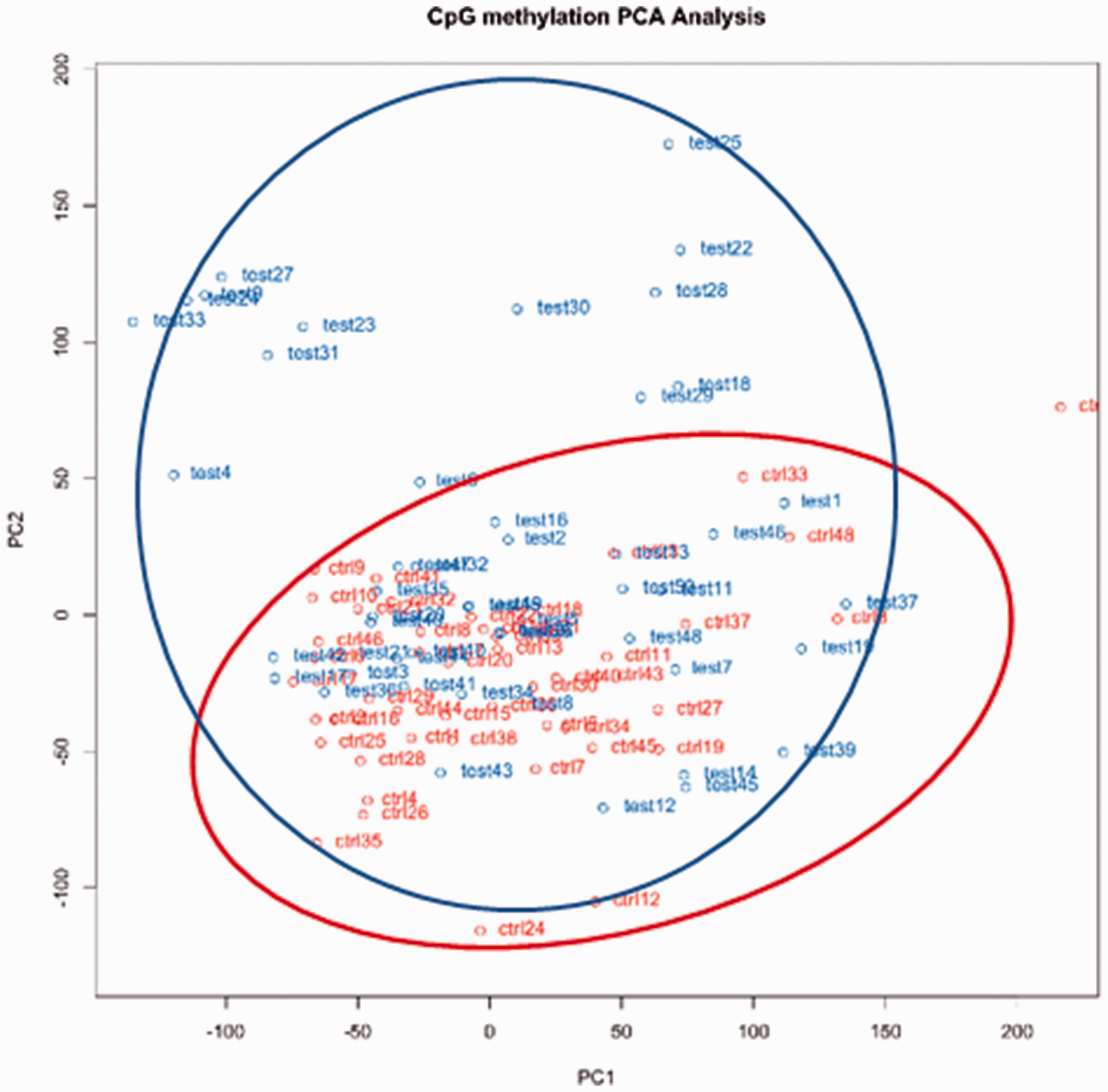
Principal component analysis of the variance between cLBP and PFCc. Notes: cLBP cases are shown in blue and PFC are shown in red.
Characterizations of differentially methylated regions associated with cLBP
Our goal was to identify differentially methylated patterns associated with cLBP. We obtained over 213k DMRs mapped to the reference human genome (hg19). At p < 0.05, we identified 65,261 (28,325 hypermethylated and hypomethylated 36,936) CpG sites. After correcting for multiple testing, we identified 159 DMRs with q-values less than 0.01 and percent methylation difference of larger than 10. The top 20 DMRs sites are shown in Table 2.
Top 20 differentially methylated regions.

Notes: Chr = chromosome.
Genomic distribution of the identified DMRs
To explore the potential functional impact of cLBP related DMRs on gene expression, we annotated the putative DMRs to predetermined genomic features. Figure 3 shows the chromosomal distribution of the annotated DMRs. The majority of the DMRs were located on chromosome 1 (8.5%), followed by chromosome 19 (7.5%) and chromosome 2 (7.2%).

Chromosomal distribution of DMRs.
As shown in Figure 4, about half of the identified DMRs were located in promoter regions (48%), with about 7% exonic regions.

Genomic region distribution of putative DMRs.
Figure 5 depicts the differential methylation annotations of the CpGs. As expected, about half of the annotated CpGs occur in CpG islands.

Genomic feature distribution of putative CpGs.
Functional genomic analyses
The majority (69.5%) of the differentially methylated CpGs were identified in protein-coding genes. To determine whether the overall pattern of differentially methylated genes was linked to cLBP disease state, we performed functional enrichment analysis using Gene Ontology. Pathway enrichment analysis revealed that cLBP-related DMRs were significantly enriched in about 165 biological processes in humans (p < 0.05). Table 3 shows biological processes that are over-represented in the DMRs with an enrichment fold change of >100. These processes affect critical physiological functions such as immune function, chondrocyte maturation, and bone mineralization.
Functional Enrichment pathways in cLBP.

Discussion
Worldwide, cLBP is a significant public health problem, and this study is among the first to evaluate DNA methylation profile associations in adults with and without cLBP. Using RRBS, we identified 159 DMRs (q < 0.01 and methylation difference > 10%), which annotated to important protein-coding genes, including CELSR1, KIF11, MINK1, and NAV1. Most of these genes were hypomethylated in individuals in cLBP with the notable exception of CELSR1, MFCE8, and MINK1. Hypomethylation in the promoter region is associated with gene expression, and hypermethylation in the gene body is associated with gene silencing. 40 Among individuals with cLBP, our findings suggest increased expression of NAV1 and KIF11 genes, regulating neuronal development.41–43 The NAV1 gene is expressed in the nervous system and plays a role in neuronal migration, 42 while the KIF11 affects synaptic transmission by regulating microtubular and axonal growth. 41 While it remains unclear how increased expression of NAV1 and KIF11 affects cLBP, it is possible that these genes’ epigenetic change may affect the development and transmission of nociceptors. Other genes that encode proteins involved in cellular activities such as cell growth, migration, and skeletal rearrangement are the CELSR1 and MINK1. Genetic alterations of CELSR1 are associated with neuronal tube defects, 44 and epigenetic regulation of CELSR1 has been associated with rheumatoid arthritis. 45 MINK1 is highly expressed during endochondral differentiation, and studies of MINK1 knock out mice have shown that MINK1 deficiency may play a role in both age- and injury-related osteoarthritis. 46 We found that the intronic regions of CELSR1 and MINK1 genes are hypermethylated in the cLBP group compared to healthy controls. Further analyses revealed that many genes in the top twenty DMRs (e.g. MFGE8, WDR5, PAX5, and DAD1) were hypomethylated, and some have previously been associated with various immune processes.47–50 However, the interpretation of the methylation status of these non-promoter regions is less straight forward.
Functional pathway analysis allows for identifying biological processes, diseases, or signaling pathways associated with groups of related genes that are altered in case samples compared to controls. 51 A functional pathway analysis revealed that the top differentially enriched biological processes in individuals with cLBP included immune signaling, endochondral ossification, and G-protein coupled receptor signaling. Our findings suggest that alterations in immune signaling pathways such as fractalkine production, T-helper 1 cell lineage commitment, N-terminal peptidyl-glycine N-myristoylation, and localization of protein to lysosome are differentially enriched in individuals with cLBP. Fractalkine (CX3C) is one of the most expressed membrane-bound chemokines in the central nervous system. 52 It is also expressed on endothelial activated proinflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) and interleukin-1, where it acts as a chemo-attractant. Emerging evidence suggests that fractalkine/CX3C is involved in the pathogenesis of chronic painful conditions such as rheumatoid arthritis and osteoarthritis.52–54 Besides fractalkine production, our results also suggest a differential commitment of genes regulating T-helper 1 cell lineage, which promote immunological processes may play a role in cLBP. Changes in T-helper 1 cell lineage have previosly been associated with arthritis. 55 In addition, epigenetic mechanisms play a role in the T helper 1 cell-lineage specific gene expression, including T-box transcription factor 21 (TBX21) and interferon γ (IFNG). Other investigators have previously reported that variations in the expression of TBX21 and IFNG genes correlate with various chronic pain conditions.56–58 These findings are consistent with previous studies that found chemokines, cytokines, and other proinflammatory genes to be differentially expressed in acute and chronic low back pain.59,60
The vertebral column is formed through chondrocyte differentiation and bone mineralization. Our study found that pathways associated with negative regulation of chondrocyte development and bone mineralization are differentially enriched in individuals with cLBP. Chondrocytes undergo sequential differentiation, proliferation, and maturation to establish future bones. 61 It has been reported that among individuals with osteoarthritis, the chondrocytes undergo degradation in response to mechanical or proinflammatory injury. This degradation process has been described as “dedifferentiation” or “transdifferentiation” because rather than merely breaking down the cartilage, the chondrocytes undergo phenotypic changes that correlate with the onset of osteoarthritis.62,63 Also, osteoarthritis may be induced by injury (mechanical, inflammation, or aging) that causes dysregulation of undifferentiated articular chondrocyte progenitors, and associated changes in endochondral ossification. 63 Thus, it is possible that non-specific cLBP may be related to epigenetic modifications that alter chondrocyte-to-osteoblast ossification. Tajerian and colleagues have previously reported that differential methylation of the gene that encodes the extracellular matric protein SPARC was associated with low back pain severity in humans and animal models. 29 In our sample, the SPARC gene was slightly hypermethylated in individuals with cLBP. This suggests that epigenetic changes to the matrix of the vertebral column may play a role in cLBP.
Genes related to the G-protein coupled receptor signaling pathway were also differentially enriched in our sample. The enriched pathways included positive regulation of synaptic cholinergic transmission, and bombesin receptor signaling. Of these pathways, the regulation of cholinergic impulses and bombesin receptor have previously been associated with nociception.64–66 Pain is modulated via cholinergic impulses at spinal and supraspinal sites, including the dorsal horn, hippocampus, and locus coerulus.67,68 Within this system, several genes (most notably the tachykinin precursor 1 [TAC1] gene, which encodes substance P and other products of the tachykinin peptide hormone family) play a role in pain modulation. 69 Expression of TAC1 genes in the locus coeruleus has been associated with chronic inflammation, 70 while differential expression in the periaqueductal gray has been associated with chronic itching. 71 Similarly, differential expression of supraspinal bomberin receptor signaling has also been associated with itching (hypersensitivity) and antinociception. 64
Several pathways were differentially enriched without known specific links to pain or nociception, including negative regulation of gastric emptying, and negative regulation of retinal cell programmed cell deaths. It is possible that these pathways contain genes that may affect nociception and pain perception.
Finally, epigenetic modifications such as DNA methylation reflect gene-environment interactions that affect gene expression without changing the underlying DNA sequence. 40 These environmental factors occur over an individual’s lifespan and may influence normal physiology and disease risk, including chronic pain. 72 Given the differential enrichment of gene involved in immune signaling and ossification, it is possible that the observed differential methylation in the cLBP group, may be related to environmental factors that promote inflammation and decrease bone formation/healing such as chronic stress and depression.18,73,74 Studies have shown that increased psychological stress may increase inflammation and alter endochondral ossification. 75 Future studies should longitudinally examine gene-environment interactions and associated DNA methylation changes in individuals in cLBP.
Strengths and limitations
The current study had several strengths, including the largest RRBS sample to date in research on non-specific cLBP. Nonetheless, our findings must be considered in the context of several limitations. First, since our study is cross-sectional, we cannot determine whether observed DNA methylation changes in the epigenome of individuals with cLBP are a consequence of the disease, or a part of its etiology. By including a general healthy sample of PFCs, we guarded against differential methylation being just a consequence of co-morbidities. Second, the sample size of our study is relatively small for genome-wide RRBS, even if it is the largest to date among cLBP methylation studies. 29 Third, our genomic DNA was extracted from peripheral blood cells, and thus our DNA methylation analysis contains a mix of cell types. However, several studies have shown that blood DNA methylation correlates strongly with brain tissue methylation,76,77 and other investigators have used blood to study DNA methylation in chronic pain conditions.15,23,26,78,79 Finally, we use computational analyses to evaluate functional pathways associated with the observed differential methylation between cLBP and PFC. Future more extensive studies should analyze actual differential gene expression (transcriptomics) and protein levels.
Conclusion
Effective management of cLBP, one of the leading causes of lives lived with disability, requires an understanding of underlying mechanisms that cause cLBP. The current study aimed to provide a better understanding of the relationship between DNA methylation modifications and cLBP. The findings are consistent with other studies suggesting a potential role of inflammation, and vertebral matrix integrity. Our study further suggests that differential methylation of genes involved in G-protein coupled cholinergic signaling correlate with cLBP. Recently targeted interventions to reverse epigenetic modifications provide hope for the development of novel effective interventions to manage non-specific cLBP.
Footnotes
Acknowledgment
We are grateful to our volunteers and the ERASED study team.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the American Association of Nurse Anesthetists Foundation Post-Doctoral Fellowship Award (2018-FS-4) to ENA, The University of Alabama at Birmingham Dean’s Scholar Award to ENA, and the National Institutes of Health grant (R01MD010441) to BRG.