
Abstract
Cav3 channels play an important role in modulating chronic pain. However, less is known about the functional changes of Cav3 channels in superficial spinal dorsal horn in neuropathic pain states. Here, we examined the effect of partial sciatic nerve ligation (PSNL) on either expression or electrophysiological properties of Cav3 channels in superficial spinal dorsal horn. Our in vivo studies showed that the blockers of Cav3 channels robustly alleviated PSNL-induced mechanical allodynia and thermal hyperalgesia, which lasted at least 14 days following PSNL. Meanwhile, PSNL triggered an increase in both mRNA and protein levels of Cav3.2 but not Cav3.1 or Cav3.3 in rats. However, in Cav3.2 knockout mice, PSNL predominantly attenuated mechanical allodynia but not thermal hyperalgesia. In addition, the results of whole-cell patch-clamp recordings showed that both the overall proportion of Cav3 current-expressing neurons and the Cav3 current density in individual neurons were elevated in spinal lamina II neurons from PSNL rats, which could not be recapitulated in Cav3.2 knockout mice. Altogether, our findings reveal that the elevated functional Cav3.2 channels in superficial spinal dorsal horn may contribute to the mechanical allodynia in PSNL-induced neuropathic pain model.
Keywords
Introduction
Peripheral nerve injury results in functional and structural plasticity, which is crucial for the pathogenesis of pain.1–3 A variety of channels in neural circuits of pain are involved in neuropathic pain. 4 Low-voltage-activated calcium channels, also referred as T-type calcium (Cav3) channels, are widely distributed in the nervous system and consist of three subtypes based on the pore-forming α1 subunit: Cav3.1 (a1G), Cav3.2 (a1H), and Cav3.3 (a1I).5,6 Since Cav3 channels can control the neuronal low-threshold spiking and exocytosis, 7 they have been reported to play a crucial role in numerous painful pathological conditions, including complete Freund’s adjuvant- or formalin-induced inflammatory pain,8,9 chronic constriction injury (CCI)- or spinal nerve ligation (SNL)-induced neuropathic pain10,11 as well as painful diabetic neuropathy (PDN),12,13 and so on. Most of the studies above are focused on the alterations of Cav3 channels in dorsal root ganglia (DRG). However, the central mechanism of Cav3 channels involved in pathological pain has not been fully elucidated so far.
Lamina II (substantia gelatinosa (SG)) neurons in the superficial spinal dorsal horn (SDH) are the second-order sensory neurons, which primarily receive and modulate nociceptive inputs from the myelinated Aδ and unmyelinated C fibers. 14 Therefore, the increasing excitatory process of SG neuron is one of the most important mechanisms for the central sensitization of pathological pain. 15 Recently, a Cav3.2-dependent link of SG neuronal excitability and cutaneous transient receptor potential vanilloid-1 (TRPV1) expressing nociceptors has been demonstrated. 16 Conditioning stimulation of optogenetically targeted cutaneous TRPV1 expressing nociceptors could evoke an increase in C-fiber activity, which may, in turn, lead to an enhanced presynaptic excitatory neurotransmission in SG neurons. 16 Moreover, either Ni2+ (a blocker of Cav3 channels) or Tat-3.2-III-IV peptide (a blocker of USP5-Cav3.2 association) could decrease the frequency of excitatory postsynaptic currents in SG neurons, strongly suggesting a role of Cav3.2 in the sensitization of the pain pathway mentioned above.16,17 Therefore, the potentiation of presynaptic Cav3.2 channels may contribute to the hyperexcitability of SG neurons, accompanying with the promotion of chronic pain. On the other hand, one previous study using a knockin strategy by inserting a green fluorescent protein tag into Cav3.2 locus, and transmission electron microscopy analyses have verified the presence of Cav3.2 in the postsynaptic area of lamina II–III, which showed higher intense labeling compared to the presynaptic zone. 18 Consistent with this study, we recently reported that Cav3 channel-mediated currents (IT) can be recorded in about half of the SG neurons by using whole-cell patch-clamp technique. 19 Although it has been shown that the expression of Cav3 channels in SDH was upregulated after nerve ligation- or paclitaxel-treatment,11,20–22 the functional changes of Cav3 channels under the neuropathic pain state have not been studied yet.
In our attempt to unravel the role of postsynaptic Cav3 channels in neuropathic pain, we investigated the changes of Cav3 channels in superficial SDH in response to partial sciatic nerve ligation (PSNL) treatment. We confirmed that the blockade of Cav3.2 channels could significantly alleviate PSNL-induced neuropathic pain. Also, we found that PSNL increased both the proportion and current density of IT without affecting its gating properties. Therefore, these data for the first time demonstrated that the augment of functional Cav3.2 channels in superficial SDH neurons may underlie the central mechanisms of mechanical allodynia in PSNL-induced neuropathic pain.
Materials and methods
Animals
Sprague-Dawley (SD) rats (150–250 g) and wild-type (WT) C57/BL6 mice (20–30 g) were obtained from the Animal Center of Nanchang University. Cav3.2 knockout (KO) mice (C57/BL6, 20–30 g) were purchased from the Jackson Laboratory (Stock No: 013770, Bar Harbor, ME, USA). Animals were housed in plastic cages with controlled room temperature (22°C–25°C) and maintained on a 12:12-h light–dark cycle. Food and water were available ad libitum. Efforts were made to minimize animal suffering as well as the number of animals. Animals of either sex were randomized to each experimental group. All animal procedures were in accordance with the protocols approved by the Institutional Animal Care and Use Committee of Nanchang University (No. 2017009) and the International Association for the Study of Pain Guidelines.
PSNL-induced neuropathy
Chronic neuropathic pain was induced by PSNL as described previously.23,24 Briefly, the right sciatic nerve was exposed at the high-thigh level under the anesthetization of 2% isoflurane. The dorsal 1/3 to 1/2 of the nerve was tightly ligated with 7-0 silk suture. For sham-operated rats or mice, the sciatic nerve was exposed without ligation.
Behavioral tests
Mechanical allodynia was measured using the electronic von Frey Aesthesiometer (rats: 38450, Ugo Basile, Italy; mice: BIO-EVF4 S, Bioseb, France). Animals were placed in Plexiglas chambers (10 × 20 × 13 cm) equipped with wire-net floor, 30 cm above the bench. An increasing force ranging from 0 to 50 g was applied to the mid-plantar area of the ipsilateral hind paw. The paw withdrawal threshold (PWT) was defined as the minimum pressure (g) required to elicit a robust and immediate withdrawal reflex of the paw. Measurements were repeated three times with an interval of 1 min. The average value of these measurements was used to determine the threshold.
Thermal hyperalgesia was measured with a plantar test apparatus (37370, Ugo Basile, Italy). Animals were kept in testing chambers (10 × 20 × 13 cm) placed on top of a grid floor. A movable infrared radiant heat source (IR = 30) was focused on the plantar surface of the hind paw until a withdrawal response occurred and the paw withdrawal latency (PWL) was recorded (in seconds). The device was set at a cutoff time of 20 s to prevent possible tissue injury. Three measurements were taken with a 5-min interval for each trial, and the mean value was used for the analysis.
Behavioral tests were performed before the operation, and at day 1, 3, 7, 10, and 14 after the operation in a blinded manner. Animals were accustomed to the Plexiglas chambers three days before performing the test and a 30 min acclimatization period at each day’s test. For those rats received intraperitoneally injection (i.p.) of ascorbic acid or saline, behavioral tests were performed at 1 h after injection on the time points mentioned above.
Intrathecal catheter implantation and intraperitoneal injection
As previously described,20,22 implantation was conducted immediately after the PSNL operation of rats. After a laminectomy at the level of L5–L6, a plastic PE-10 tube (outer diameter: 0.5 mm, inner diameter: 0.25 mm; AniLab Co. Ltd, China) filled with saline was inserted into the intrathecal space and advanced rostrally about 2 cm. Correct position of the catheter tip was confirmed by a characteristic tail-flick response. An osmotic minipump with a flow rate of 0.25 µl/h for 14 days (Model 1002, ALZET, USA) was attached to the tip of the tube. The catheter and the pump were then secured subcutaneously in the dorsal skin. Those representing any dysfunction of motor were sacrificed immediately after recovery from anesthesia. The secondary verification of the catheter tip was checked at day 14 after PSNL operation. Any rats exhibited incorrect position of the tip would be excluded from final analysis. NiCl2 (0.5 µg/h) and TTA-A2 ([2-(4-cyclopropylphenyl)-N-((1R)-1-{5-[(2,2,2-trifluoroethyl)oxo]-pyridin-2-yl}ethyl)acetamide], 0.35 µg/h) were continuously infused intrathecally (i.t.) starting from the first day of PSNL operation. Saline at a flow rate of 0.25 µl/h was used as the control. Ascorbic acid was freshly diluted with saline and was given to the rats at the dose of 0.5 g/kg (in a volume of 2 ml, i.p.) at 1 h before the operation 25 and daily for 14 days after operation. Equivalent volume of saline was used as the control.
Quantitative real-time polymerase chain reaction
Ipsilateral superficial SDH at the level of L4–L5 was collected from sham and PSNL rats under deep anesthesia with urethane (1.5 g/kg, i.p.) at 14 days after operation. Total RNA was extracted using RNAiso™ Plus (9108; Takara Bio, Kyoto, Japan) according to the manufacturer’s protocols. Reverse transcription (37°C for 15 min and 85°C for 5 s) from total RNA (0.5 µg) was carried out with an ABI Veriti PCR machine (Applied Biosystems, Forster City, CA, USA). cDNA was synthesized using Primescript™ RT reagent kit (RR037A; Takara Bio) and stored at −20°C until use. Reaction of quantitative real-time polymerase chain reaction (qRT-PCR) for each gene was performed in triplicate using the SYBR® Premix Ex Taq™ II kit (RR820A; Takara Bio) on a StepOnePlus thermocycler (Applied Biosystems). The cycling conditions were as follows: a cycle of 95°C for 30 s, followed by 40 cycles at 95°C for 5 s and at 60°C for 30 s. Expression levels of mRNA were normalized to β-actin and quantified using the 2−ΔΔCT method.26,27 Reactions without any template were used as negative controls. The specific forward and reverse primers are shown in Table 1.
Forward and reverse primers of Cav3.1, Cav3.2, Cav3.3, and β-actin.

Western blot
The fresh tissue samples stated above were homogenized and lysed in lysis buffer containing 0.5% lubrol-px, 50 mM KCl, 2 mM CaCl2, 20% glycerol, 50 mM Tris-HCl (pH = 7.4), 1 mM NaN3, and a mixture of 1% protease inhibitor (P8340; Sigma-Aldrich, St. Louis, MO, USA) for 1 h on ice followed by centrifugation (5415R; Eppendorf, Germany) at 12,000 relative centrifugal force for 15 min at 4°C. For western blot assays, samples (200 µg protein for Cav3 and 40 µg protein for β-actin) were loaded onto 6% to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (P1200; Solarbio, China) and then transferred to nitrocellulose membranes (HATF00010; Millipore, USA). Membranes were incubated with rabbit polyclonal anti-Cav3 (1:200; Alomone Labs, Israel) and rabbit polyclonal anti-β-actin (1:1000; Proteintech, IL, USA) antibodies overnight at 4°C. Afterward, membranes were incubated with goat anti-rabbit IgG secondary antibody (1:1000; Thermo Fisher Scientific, USA) for 6 h at 4°C. The band intensity of the detected proteins was determined by Image J software (NIH Image analysis website: http://rsb.info.nih.gov/ij/). β-actin was used to normalize the quantification.
Whole-cell patch-clamp recordings
Spinal cord slices were prepared as our previous studies.19,28 In brief, animals were deeply anesthetized with urethane (1.5 g/kg, i.p.) and were perfused transcardially with ice-cold carbonated (95% O2 and 5% CO2) dissection solution containing (in mM) 240 sucrose, 2.5 KCl, 3.5 MgCl2, 0.5 CaCl2, 1.25 NaH2PO4, 0.4 ascorbic acid, 2 sodium pyruvate, 25 NaHCO3, and 1 kynurenic acid. The lumbosacral sections were immediately dissected and immersed in the same solution. Animals were then sacrificed by exsanguination after extraction while still under anesthesia. Transverse slices measuring 400–450 µm in thickness were cut with a vibratome (VT1000S; Leica, Nussloch, Germany) and equilibrated in artificial cerebrospinal fluid solution (aCSF) at 32°C for at least 30 min before recording. The aCSF contained (in mM) 117 NaCl, 3.6 KCl, 2.5 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 25 NaHCO3, 11 D-glucose, and 2 sodium pyruvate.
After incubation, one slice was mounted on a submerged recording chamber and continuously perfused with aCSF (2–4 ml/min) at room temperature (23°C–25°C). SG neurons were visualized with an IR-DIC camera (IR-1000; Dage, Michigan City, IN, USA). Recording electrodes were pulled from borosilicate glass (World Precision Instruments, Sarasota, FL, USA) with a micropipette puller (P-97; Sutter Instrument, USA). Typical resistance of the pipette was 3–6 MΩ when filled with an intracellular solution containing the following (in mM): 92 CsMeSO4, 43 CsCl, 10 phosphocreatine, 0.5 EGTA, 10 HEPES, 4 Mg-ATP, 0.3 Li-GTP, and 5 tetraethylammonium chloride (pH = 7.2 adjusted with CsOH, 300 mOsm). To isolate Cav3 currents, 1 µM tetrodotoxin (TTX) was added to the external solution (aSCF) during recording. Signals were amplified with an EPC-10 amplifier and Patchmaster software (HEKA Electronik, Lambrecht, Germany). Series resistances were generally 10–30 MΩ and were monitored throughout the recording period. Data were excluded if the series resistance changed by >20%.
Chemicals
TTA-A2 and TTX were purchased from Alomone Labs and Tocris Bioscience (Bristol, UK), respectively; other chemicals were from Sigma-Aldrich. All drugs were dissolved in distilled water for in vitro experiments or in saline for in vivo experiments, except for TTA-A2, which was dissolved in dimethyl sulfoxide at high times first and then diluted to the final concentration in saline.
Statistical analysis
Data are represented as mean ± standard error of the mean. Curve fitting was performed using GraphPad Prism 7.0 (GraphPad Software, USA). SPSS version 17.0 (SPSS Inc., Chicago, IL, USA) was used for statistical analysis. Shapiro–Wilk test was used to assess the normal distribution of data. Levene’s test was used to test the homogeneity of variance. Student’s t test (paired or unpaired, as appropriate) or Mann–Whiteny U was used for the comparison of the mean values between two groups. Two-way analysis of variance with post hoc of Bonferroni test or Dunnett’s test was used for multiple comparisons. Chi-square test was used to determine the difference of the proportion of IT in SG neurons. Significance was set at p < 0.05.
Results
Cav3 channel blockers alleviated PSNL-induced mechanical allodynia and thermal hyperalgesia in rats
PSNL was used as the neuropathic pain model in this study. As shown in Figure 1(a), basal responses (preoperation) to von Frey Aesthesiometer were similar between sham and PSNL rats, with a mean PWT of 25.8 ± 0.9 g and 26.8 ± 0.9 g (p > 0.05), respectively. However, PWT was significantly decreased at postoperation day (POD) 1 (p < 0.001), 3 (p < 0.01), 7 (p < 0.01), 10 (p < 0.001), and 14 (p < 0.001) in PSNL rats compared to those of the sham rats. Similar results were found in the thermal hyperalgesia test (Figure 1(b)). In PSNL rats, the baseline of PWL was 10.7 ± 0.3 s, which was largely decreased to 7.1 ± 0.4 s at POD 1 (p < 0.001) and remained at lower levels until POD 14 (p < 0.01–0.001). Together, these results confirm that PSNL rats developed neuropathic pain behaviors starting from POD 1 and persisting to POD 14.
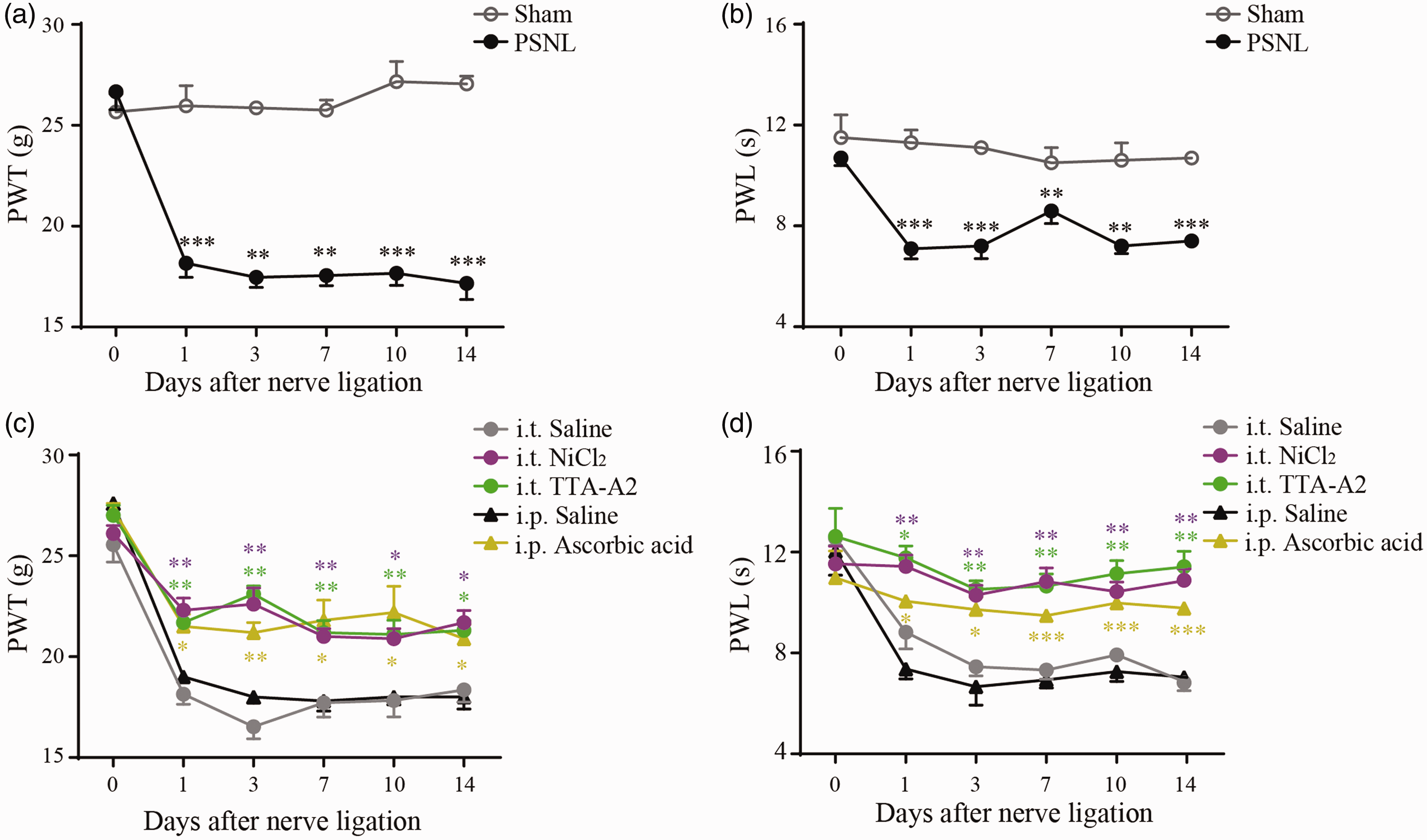
Analgesic effects of Cav3 channel blockers on rat neuropathic pain model induced by PSNL. (a) and (b) Time courses of mechanical allodynia and thermal hyperalgesia of sham (n = 6) and PSNL (n = 6) rats over a period of 14 days after operation. (c) and (d) Time courses of mechanical allodynia and thermal hyperalgesia of rat PSNL models treated with NiCl2 (0.5 µg/h, i.t., n = 6), TTA-A2 (0.35 µg/h, i.t., n = 7), and ascorbic acid (0.5 g/kg, i.p., n = 5). Intrathecal (n = 5) or intraperitoneal (n = 3) injection of saline was used as the control. *p < 0.05, **p < 0.01, ***p < 0.001, compared with the control. PSNL: partial sciatic nerve ligation; PWT: paw withdrawal threshold; PWL: paw withdrawal latency; i.t.: intrathecal injection; i.p.: intraperitoneal injection; TTA-A2: [2-(4-cyclopropylphenyl)-N-((1R)-1-{5-[(2,2,2-trifluoroethyl)oxo]-pyridin-2-yl}ethyl)acetamide].
Next, we tested whether Cav3 channel blockers could alleviate PSNL-induced hypersensitivity. Compared with PSNL saline control, NiCl2 (a classical Cav3 blocker) greatly suppressed the mechanical allodynia and thermal hyperalgesia over the experimental period (p < 0.05, Figure 1(c) and (d)). To exclude the possible inhibitory effect of NiCl2 on R-type calcium channels, 29 we also examined the effects of TTA-A2 (a specific Cav3 blocker) and ascorbic acid (a blocker selectively inhibits Cav3.2 among the three Cav3 subtypes) on PSNL rats as well. In fact, along with the result of NiCl2, both TTA-A2 and ascorbic acid profoundly relieved the pain behavioral responses of PSNL rats (p < 0.05, Figure 1(c) and (d)). Collectively, these data show that blockade of Cav3 channels can repress PSNL-induced neuropathic pain.
PSNL increased the expression of Cav3.2 in rat superficial SDH
Although Cav3.2 was the primary subtype involved in chronic pain, 10 Cav3.1 and Cav3.3 may also possess a role in pain modulation.30,31 Therefore, we next investigated whether PSNL could induce changes in Cav3 mRNA and protein levels in rat superficial SDH. As shown in Figure 2(a), Cav3.2 mRNA increased 1.30 ± 0.09 times in PSNL rats as compared to that of the sham rats (p < 0.05). However, no significant difference was found in mRNA level of Cav3.1 and Cav3.3 between two groups, which were 1.20 ± 0.09 and 1.20 ± 0.14 times of control, respectively (both p > 0.05). Consistently, western blot analysis showed that the protein level of Cav3.2 increased 1.90 ± 0.26 times after PSNL (p < 0.05), while there was no marked change for Cav3.1 (1.11 ± 0.19 times of control) and Cav3.3 (1.12 ± 0.14 times of control) (both p > 0.05, Figure 2(b) and (c)). Figure 2(d) illustrates that the Cav3.2 band at around 250 kDa was seen in WT mice but was absent in Cav3.2 KO mice, validating the specificity of the Cav3.2 antibody used in the current study. Taken together, these data suggest that PSNL increased the expression of Cav3.2, but not Cav3.1 and Cav3.3, in rat superficial SDH.

Expression of Cav3.1–3.3 in superficial SDH in sham and PSNL rats. (a) qRT-PCR results of Cav3 mRNA expression in superficial SDH relative to β-actin in sham (n = 6) and PSNL (n = 6) rats. (b) and (c) Representative western blot (b) and the quantitative analysis (c) of Cav3 protein obtained from the superficial SDH of sham (n = 6) and PSNL (n = 6) rats. The molecular weights of Cav3 and β-actin were ∼250 and ∼42 kDa, respectively. (d) Representative western blot of Cav3.2 from WT and KO mice. *p < 0.05, compared with sham control. PSNL: partial sciatic nerve ligation; S: sham; P: PSNL; WT: wild-type; KO: knockout.
Our and others’ studies identified that Cav3.2 is essential for pain signaling; 10 thus, we next explored the PSNL-induced hypersensitivity in Cav3.2 KO and WT mice. The basal PWT and PWL of Cav3.2 KO mice were not apparently different from those of the WT mice (p > 0.05, Figure 3(a) and (b)). Moreover, either Cav3.2 KO or WT mice developed a mechanical allodynia and thermal hyperalgesia for up to 14 days following PSNL. However, the mechanical allodynia in Cav3.2 KO mice was partly suppressed as shown by an increase in PWT after PSNL compared to WT-PNSL mice (p < 0.05, Figure 3(a)). However, no significant difference was observed in PWL between the Cav3.2 KO- and WT-PSNL mice (p > 0.05, Figure 3(b)). In accordance with the results illustrated in Figure 1, these data demonstrate that deletion of Cav3.2 may alleviate the PSNL-induced mechanical allodynia but not thermal hyperalgesia.

PSNL-induced mechanical allodynia and thermal hyperalgesia in WT and Cav3.2 KO mice. (a) and (b) Time courses of mechanical allodynia and thermal hyperalgesia of sham and PSNL mice over a period of 14 days after operation (WT-sham: n = 4, WT-PSNL: n = 4; KO-sham: n = 9, and KO-PSNL: n = 10). *p < 0.05, **p < 0.01, ***p < 0.001, compared with sham. #p < 0.05, ##p < 0.01, compared with KO-PSNL mice. PSNL: partial sciatic nerve ligation; WT: wild-type; KO: knockout; PWT: paw withdrawal threshold; PWL: paw withdrawal latency.
PSNL enhanced functional Cav3 channel activities in rat SG neurons
Above data of qRT-PCR and western blot analysis verified that PSNL upregulated the expression of Cav3.2 in superficial SDH. However, whether and how PSNL could affect IT in SG neurons remain unknown. Our recent study showed that IT was observed in 44.5% of SG neurons in normal SD rats. 19 In this study, we further found that IT was recorded in 41 of the 69 neurons (59.4%) in sham rats. As expected, this percentage was dramatically increased to 76.7% (46 of the 60 neurons, p < 0.05, Figure 4(a)) in PSNL rats. Moreover, the peak amplitude of IT which was activated by a 200-ms voltage step to −50 mV from a holding potential (Vhold) of −110 mV for 500 ms was largely increased from −78.8 ± 12.3 pA to −124.3 ± 14.9 pA (p < 0.05, Figure 4(b)) in sham and PSNL rats, respectively. This indicates that PSNL led to a 57.7% increase in IT amplitude compared to sham control. Then, we studied the kinetics of Cav3 currents in SG neurons from PSNL and sham rats. Figure 4(c) to (e) illustrated that the current density of IT was larger in PSNL rats than sham control at the potential of −85 to −110 mV (p < 0.05). Next, we traced the activation and inactivation curves but did not find any significant change of PSNL on the kinetic properties of IT (Figure 4(f)). The fitting parameters, V50 (half-activation or half-inactivation potentials) and k (slope factor), of sham and PSNL rats were compared with each other. The fitting parameters of activation and inactivation curves were not significantly altered after PSNL (p > 0.05, Figure 4(g) and Table 2 (control)). These electrophysiological data combined with the results of qRT-PCR and western blot suggest that PSNL increased the proportion of functional Cav3.2 channels in SG neurons.

The proportion and electrophysiological characteristics of Cav3 currents in SG neurons of PSNL rats. (a) The proportion of neurons with or without IT in sham (41/69, 59.4%) and PSNL rats (46/60, 76.7%). (b) Peak amplitudes of IT (Vhold = −110 mV) from sham (n = 41) and PSNL (n = 46) rats. (c) Current density (peak current amplitude divided by cell’s capacitance) against test potential of IT from sham (n = 41) and PSNL (n = 46) rats. (d) and (e) Representative activation and inactivation traces of IT in SG neurons from sham and PSNL rats. Activation currents (d) were evoked by the voltage steps from −110 mV (Vhold) to test potentials from −70 mV through −40 mV in a 5-mV step. Inactivation currents (e) were evoked by test steps to −50 mV after a 0.5-s prepulse at potentials from −60 mV to −110 mV in a 5-mV step. (f) Steady-state activation and inactivation of IT in the sham and PSNL rats. (g) Bar graphs of half-activation or half-inactivation potentials of IT (activation: sham, n = 11, PSNL, n = 16; inactivation: sham, n = 14, PSNL, n = 22). *p < 0.05, compared with sham control. PSNL: partial sciatic nerve ligation.
Fitting parameters of activation and inactivation curves.

PSNL: partial sciatic nerve ligation; V50: half-activation or half-inactivation potentials; k: slope factor.
Effects of PSNL on Cav3 currents in SG neurons from Cav3.2 KO mice
If the functionally potentiated IT following PSNL is mainly caused by Cav3.2 subtype, then this facilitation might be diminished in Cav3.2 KO mice. To test this hypothesis, we recorded IT from SG neurons of sham and PSNL Cav3.2 KO mice. As predicted, unlike the findings in SD rats, the proportion of neurons with IT was not significantly different between sham and PSNL mice, which were 58.3% (21/36) and 56.8% (25/44) (p > 0.05, Figure 5(a)), respectively. The mean IT amplitudes were −50.1 ± 8.8 pA and −55.2 ± 7.1 pA (Vhold = −110 mV) in sham and PSNL mice (p > 0.05, Figure 5(b)), respectively. Moreover, no change of current density was found between the two groups (p > 0.05, Figure 5(c)).

Cav3 currents in SG neurons of Cav3.2 KO mice. (a) The proportion of neurons with or without IT in sham and PSNL mice. (b) Peak amplitudes of IT (Vhold = −110 mV) from sham and PSNL KO mice. (c) Current density against test potential of IT from sham and PSNL mice. Sham, n = 21; PSNL, n = 25 (p > 0.05). PSNL: partial sciatic nerve ligation.
NiCl2 blocked Cav3 currents in SG neurons from sham and PSNL rats with similar potency
Our data reveal that Cav3 channel blockers could suppress the PSNL-induced mechanical allodynia and thermal hyperalgesia. However, whether the magnitude of the blockade on IT is the same in PSNL and sham control rats is not clear. As shown in Figure 6(a) and (b), bath application of NiCl2 (100 µM) for 15 min decreased IT amplitude in both PSNL and sham rats (p < 0.05). Meanwhile, the inhibitory ratio was not significantly different between the sham (45%) and the PSNL groups (57%) at −45 mV (p > 0.05) in which IT reached its maximal amplitude (Figure 6(c)). Furthermore, we compared the effect of NiCl2 on the sham and PSNL rats. First, under the treatment of NiCl2, the activation and inactivation properties did not show significant difference between the sham and PSNL rats (p > 0.05, Figures 6(d) and (e) and Table 2). Second, those properties were not altered before and after the treatment of NiCl2 in both groups (p > 0.05, Table 2). Taken together, these results suggested that NiCl2 plays a similar effect on both normal and pathological states.

Blockade effects of Cav3 currents in SG neurons from sham and PSNL rats by NiCl2. (a) and (b) The current–voltage (I–V) curves (a) and representative traces (b) of IT from sham and PSNL rats before or after the treatment of NiCl2 (sham, n = 11; PSNL, n = 16). (c) Inhibitory ratio of NiCl2 on IT amplitudes between sham and PSNL rats at −45 mV (sham, n = 11; PSNL, n = 16). (d) Steady-state activation and inactivation of IT between sham and PSNL groups after treated with NiCl2. (e) Effects of NiCl2 on half-activation or half-inactivation potentials of Cav3 currents in sham and PSNL rats (activation: sham, n = 11, PSNL, n = 16; inactivation: sham, n = 14, PSNL, n = 22). *p < 0.05, **p < 0.01, ***p < 0.001, compared with control. PSNL: partial sciatic nerve ligation.
Discussion
In this study, the effect of PSNL on Cav3 channels in superficial SDH was investigated. We identified that it was the Cav3.2, but not Cav3.1 and Cav3.3, increased both mRNA and protein levels after PSNL treatment. Moreover, our studies as presented here provide the first line of evidence for that not only the proportion but also the current density of Cav3 currents were increased under neuropathic pain condition.
In the 1980s, Cav3 channels were reported in sensory neurons including DRG and spinal SDH neurons, indicating a possible role of Cav3 channels in modulating nociceptive information.32–36 This hypothesis was later confirmed by the development of pharmacological Cav3 channel blockers, which showed antinociceptive effects on the mechanical allodynia and thermal hyperalgesia.37,38 Consistent with these observations, our behavioral experiments as presented here showed that the admission of Cav3 channel blockers either by continuous intrathecal injection or intermittent intraperitoneal injection to PSNL rats elicit a strong and stable analgesic effect until POD 14. Also, genetic interference by either general KO31,39–41 or temporary knockdown of Cav3 with the oligonucleotide antisense (AS)10,42 or small interfering RNA43,44 alleviated the nociceptive behavior. Remarkably, among the three subtypes of Cav3 channels, Cav3.2 is the most critical subtype. Bourinet et al. found that it was the AS-Cav3.2 but not the AS-Cav3.1 and AS-Cav3.3 exhibited antihyperalgesic and antiallodynic effects in CCI rats. 10 However, in another neuropathic pain model of chronic compression of the DRG, both AS-Cav3.2 and AS-Cav3.3 led to the reverse of nociceptive behavior. 42 Moreover, Cav3.1 KO mice displayed a decreased mechanical hypersensitivity in either trigeminal neuropathic pain or SNL-induced thermal hyperalgesia.31,41 Therefore, it is possible that distinct Cav3 subtypes play a different role in various neuropathic pain models. Consistent with the aforementioned study of Bourinet et al. using qRT-PCR and western blot analysis, we found an elevated expression of Cav3.2, but not Cav3.1 and Cav3.3, in superficial SDH from PSNL rats, demonstrating that Cav3.2 is essential for PSNL-induced neuropathic pain. Moreover, we noticed that the mRNA level and the protein level of Cav3.1–3.3 are not well correlated under PSNL treatment. The reason for this discrepancy may be due to the fact that there are many complicated and varied posttranscriptional mechanisms involved in turning mRNA into protein that are not yet sufficiently well defined. Furthermore, numerous reports have concluded that protein amount cannot be deduced by measuring mRNA concentration.45,46
These findings were recapitulated by observing the nociception in Cav3.2 KO mice. In line with previous reports,24,31,40 we found that the baseline levels of PWT and PWL in Cav3.2 KO mice were not remarkably different from that of the WT mice. However, unlike the study which showed that the mechanical allodynia and thermal hyperalgesia in Cav3.2 KO mice was not differ from WT mice in SNL-induced neuropathic pain, 40 we noticed that PSNL-induced mechanical allodynia, but not thermal hyperalgesia, in Cav3.2 KO-PSNL mice was partly alleviated compared to WT-PSNL mice. Three possible mechanisms might explain these discrepancies. First, the model of neuropathic pain used was different. Although both SNL and PSNL display behavioral nociceptive responses of neuropathy, there is a considerable difference either in the magnitude of each pain component or the effect of sympathectomy. 47 Second, the methods utilized to assess the mechanical sensitivity were different. Third, noxious heat and mechanical stimuli are modulated through different pathway.48–50 For example, mechanical allodynia but not heat hyperalgesia was developed in Kv1.1 dominant-negative mice 51 or ERK2-deficient mice. 52 Further investigations are required to understand the molecular mechanisms of Cav3.2 channels underlie nerve injury-induced mechanical allodynia and thermal hyperalgesia.
Neuropathic pain-induced functional alterations of Cav3 currents recorded from DRG neurons was well-documented,30,53–57 such as the increased percentages of IT-expressing DRG neurons, the current density, and the gating parameters, and so on. Recently, the increased Cav3 current density was found in SDH neurons at seven days after SNL-treated. 11 Consistent with this study, we reported here for the first time that both the proportion and the current density of Cav3 currents were elevated in SG neurons from PSNL rats compared to that of the sham rats, without any change in the kinetics of activation and inactivation. Interestingly, for the first time, we recorded the Cav3 currents in SG neurons from Cav3.2 KO-PSNL mice and found that PSNL neither affect the proportion nor the current density of IT. Therefore, our present findings unveil a novel mechanism underlying the augmentation of nociception in PSNL-induced neuropathic pain, possibly due to the increased transcription of Cav3.2, leading to the elevated function of Cav3.2 channel. Posttranslational modifications such as glycosylation13,58,59 and deubiquitination16,17,60 play an essential role in regulating the expression and function of Cav3.2 channels. For example, glycosylation of Cav3.2 channels could increase current density in small DRG neurons from PDN models. 13 Moreover, in CCI mice, interaction of deubiquitinating enzyme USP5 and Cav3.2 was upregulated on the ipsilateral SDH tissue. 17 Meanwhile, immunostaining method also demonstrated that the expression of USP5 was increased in SDH of spared nerve injury mice. 17 On the contrary, knockdown of USP5 reduced Cav3.2 currents in Cath.a-differentiated (CAD) cells, which express USP5 endogenously. 17 Whether and how these modulations affect SG neurons under neuropathic pain conditions need to be further investigated.
When treated the neuropathic pain with Cav3 channel blockers, it is concerned that whether the inhibitory potency is the same as the physiological conditions. It has been reported that the blocked effect of NiCl2 on IT was not different from the control in CCI-injured small DRG neurons. 57 Meanwhile, another Cav3 channel blocker, mibefradil, also showed a similar effect on IT in control and streptozotocin-injured DRG neurons. 54 Consistent with these results, we did not find any significant difference for the inhibitory ratio of NiCl2 on IT in PSNL-injured SG neurons. Moreover, NiCl2 did not have any impact on the gating parameters of Cav3 channel in sham and PSNL rats. Overall, these data suggest that the molecular composition of Cav3 currents is little affected in SG neurons of PSNL rats.
In summary, our findings demonstrate an elevated expression and function of Cav3.2 channels in the superficial SDH of PSNL model, which might provide new insights into the contribution of Cav3.2 in chronic pain.
Footnotes
Author Contributions
T Liu conceptualized the study, interpreted the data, and drafted and edited the manuscript. X-J Feng performed PSNL model, i.t. and i.p. injections, qRT-PCR, western blot, and electrophysiological experiments. L-X Ma provided discussion in study design and interpretation of data. C Jiao performed the behavioral tests. H-X Kuang, F Zeng, X-Y Zhou, X-E Cheng, and M-Y Zhu assisted in the analysis of data. D-Y Zhang and C-Y Jiang supervised and supported the study. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by the National Natural Science Foundation of China (No. 31660289 to Tao Liu and 81560198 to Da-Ying Zhang) and Outstanding Young People Foundation of Jiangxi Province (20171BCB23091).