
Abstract
Accumulating evidence shows that inhibition of glycogen synthase kinase-3beta (GSK-3β) ameliorates cognitive impairments caused by a diverse array of diseases. Our previous work showed that spared nerve injury (SNI) that induces neuropathic pain causes short-term memory deficits. Here, we reported that GSK-3β activity was enhanced in hippocampus and reduced in spinal dorsal horn following SNI, and the changes persisted for at least 45 days. Repetitive applications of selective GSK-3β inhibitors (SB216763, 5 mg/kg, intraperitoneally, three times or AR-A014418, 400 ng/kg, intrathecally, seven times) prevented short-term memory deficits but did not affect neuropathic pain induced by SNI. Surprisingly, we found that the repetitive SB216763 or AR-A014418 induced a persistent pain hypersensitivity in sham animals. Mechanistically, both β-catenin and brain-derived neurotrophic factor (BDNF) were upregulated in spinal dorsal horn but downregulated in hippocampus following SNI. Injections of SB216763 prevented the BDNF downregulation in hippocampus but enhanced its upregulation in spinal dorsal horn in SNI rats. In sham rats, SB216763 upregulated both β-catenin and BDNF in spinal dorsal horn but affect neither of them in hippocampus. Finally, intravenous injection of interleukin-1beta that induces pain hypersensitivity and memory deficits mimicked the SNI-induced the differential regulation of GSK-3β/β-catenin/BDNF in spinal dorsal horn and in hippocampus. Accordingly, the prolonged opposite changes of GSK-3β activity in hippocampus and in spinal dorsal horn induced by SNI may contribute to memory deficits and neuropathic pain by differential regulation of BDNF in the two regions. GSK-3β inhibitors that treat cognitive disorders may result in a long-lasting pain hypersensitivity.
Keywords
Introduction
Chronic pain affects 20% to 30% of the world population. 1 Two thirds of the chronic pain patients suffer from memory deficits.2,3 Previous works showed that peripheral nerve injury, which induces chronic neuropathic pain, impairs working memory or short-term memory (STM) and inhibits long-term potentiation (LTP) in hippocampus, a synaptic model of memory,4,5 in rats and mice.6–9 Accordingly, dysfunction of hippocampus is responsible for this form of memory deficits. The notion is confirmed by the later works demonstrating that the hippocampus volumes in human patients with chronic back pain and complex regional pain syndrome are substantially reduced 10 and that working memory deficits in spared nerve injury (SNI) model of neuropathic pain are associated with the decrease in number of excitatory synapses in hippocampus 11 and in hippocampus-prefrontal cortex connection in rats. 12 Our recent work 13 shows that the overproduction of interleukin-1beta (IL-1β) in plasma and in the brain regions related to pain perception and memory formation is sufficient to induce behavioral signs of neuropathic pain and memory/emotional deficits following SNI. However, the downstream molecules of IL-1β remains elusive.
It is well established that activation ofBDNF/TrkB pathway is essential for both memory formation in hippocampus14,15 and the development of neuropathic pain.16,17 Interestingly, BDNF level is increased in spinal dorsal horn but decreased in hippocampus11,18,19 following peripheral nerve injury. The mechanisms underlying opposite change in BDNF expression are largely unknown.
In recent years, canonical β-catenin-dependent Wnt signaling has been proved to play important roles in both hippocampal memory and neuropathic pain. Similar to BDNF, downregulation of β-catenin, a transcription factor, in hippocampus impairs memory consolidation,20,21 while upregulation of β-catenin in spinal dorsal horn causes neuropathic pain.22,23 The level of β-catenin is regulated byGSK-3β. The activity of GSK-3β is reduced by phosphorylation of its Ser924 and enhanced by phosphorylation of Tyr216. 25 The active GSK-3β degrades β-catenin, and thus prevents its translocation to the nucleus for gene transcription. 26
Accumulating evidence shows that inhibition of GSK-3β ameliorates cognitive impairments caused by a diverse array of diseases, such as Alzheimer's disease, Down syndrome, Parkinson’s disease, spinocerebellar ataxia, and traumatic brain injury (see King et al. 27 for a review). Therefore, GSK-3β inhibitors are believed as promoting candidates for treatment of cognitive deficits. In the present work, we evaluated the roles of GSK-3β on STM and pain hypersensitivity in SNI and sham rats, and the mechanisms underlying the effects were investigated.
Materials and methods
Animals
Male Sprague-Dawley rats weighing 200 to 240 g from the Institute of Experimental Animal (Sun Yat-sen University, Guangzhou, China) were used. The rats were housed individually and free access to food and water within a room maintained on a 12/12 light/dark cycle. The environment temperature and humidity were kept at 24 ± 1°C and 50% to 60%. All animal experiment procedures were approved by the local co
mmittee and were performed in accordance with the protocol of the National Institutes of Health on animal care.
SNI model
The SNI surgery was performed as described by Decosterd and Woolf. 28 Under chloral hydrate anesthesia (0.4 g/kg, intraperitoneally (i.p.)), three peripheral branches of the sciatic nerve of the left hind limb were exposed. The common peroneal and the tibial nerves were ligated and cut (2 mm sections removed), but the sural nerve was carefully kept intact. The surgical incision was sutured in two layers.
Behavioral tests
Pain sensitivity testing was performed before and after surgery or drug administration. The animals were habituated for 15 min in separate transparent Plexiglas chambers before tests. Mechanical sensitivity was tested with the up-down method using a set of von Frey hairs (0.4–25 g, North Coast medical) as described earlier.29 . Briefly, each von Frey hair application kept a 7 s on the sural nerve innervation area in the hindpaw. A quick withdrawal or licking paw indicates a positive response. The decrease in paw withdrawal thresholds (PWTs) indicates mechanical allodynia. Heat hypersensitivity was tested using Plantar Test System (7370, UgoBasile) described previously. 30 Briefly, a radiant heat from a light source under a transparent glass floor was applied at the hindpaw plantar surface alternately with 5 min intervals between each tests. Three measurements of paw withdrawal latencies (PWLs) were averaged.
Novel object recognition test
The test was carried out to measure the STM and long-term memory of SNI and sham animals. The apparatus consisted of a round arena (diameter: 50 cm for mouse and 80 cm for rat) with black (for rat) or white (for mouse) walls and floor. The box and objects were cleaned between trials to stop the build-up of olfactory cues. Animals received two sessions of 10 min each in the empty box to habituate them to the apparatus and test room. Twenty-four hours later, each rat was first placed in the box and exposed to two identical objects for 10 min (sample phase). And then one object was replaced by a new one and the rat was placed back in the box and exposed to the familiar object and to a novel test object for a further 10-min (acquisition phase). The STM was tested 10 min after “sample phase” (10-min retention) and long-term memory 24 h after sample phase (24-h retention) on the same cohorts of animals. The experimenters measured the time spent exploring each object. The recognition index was calculated as the percentage ratio of time spent exploring the novel object over total exploration time during acquisition phase. All behavioral assays were performed by the experimenters who were blind to the design of the study.
Drugs and applications
GSK-3β inhibitors Styrene-butadiene copolymer (SB) 216763 (S3442, Sigma Aldrich) or analytical reagent (AR)-A014418 (T1881, Topscience) were dissolved in DMSO (dimethyl sulfoxide), stored as stock solutions at concentrations of 54 mM and 62 mM, respectively, at 4°C. The SB216763 was diluted with solvent sterile water and polyethylene glycol 400 (PEG 400) (1:2) and AR-A014418 with sterile water (ddH2O) to the appropriate concentrations immediately before administration. The final concentration of DMSO or PEG 400 was less than 0.2%.
SB216763 (5 mg/kg, 0.8 ml in volume) or the same volume of vehicle was i.p. injected for three times at one day before and three and six days after SNI or sham surgery. For intrathecal injection of AR-A014418, a PE-10 intrathecal catheter (BD, USA) was implanted in the lumbar enlargement (close to the L3–4 segments) five days before intrathecal drug administration. The efficacy of the embedded intrathecal catheter was verified by observing transient hindpaw paralysis induced by intrathecal delivery of lidocaine (2%, 10 μl) four days after surgery. Rats that failed to show any paralysis were excluded from the subsequent experiments. AR-A014418 (400 ng/kg, 12 μl in volume, daily) or the same volume of vehicle was injected through the catheter 30 min before and after one to six days after SNI or sham operation.
Intravenous injection of recombinant rat IL-1β
Recombinant rat IL-1β (rrIL-1β, R&D Systems) was diluted to 100 ng/ml in 0.1% bovine serum albumin in saline before administration. Our previous work showed that the peak concentration of IL-1β in plasma reaches to 800 pg/ml in SNI rats, and elevation of plasma IL-1β level to 1 ng/ml by intravenous injection of rrIL-1β (100 ng/ml, 150 μl in volume) three times per day for successive seven days in naïve rat mimics the effects of SNI, inducing mechanical allodynia and memory deficits. 13 The same manipulation was tested to induce the opposite changes in GSK3β, β-catenin and BDNF in the dorsal horn and the hippocampus.
Evans blue distribution after intrathecal injection
To determine whether the drug injected intrathecally (i.t.) is able to reach to the hippocampus, 1% Evans blue (EB) in saline (30 μl, Sigma-Aldrich) or saline with the same volume was intrathecally injected. Five hours later, the rats were perfused transcardially with 200 ml 0.1 M phosphate-buffered saline, and the spinal cord and whole brain were exposed to observe the EB distribution.
Western blotting
Rats were anesthetized, and the lumbar spinal dorsal horn segments and hippocampus ipsilateral to surgeries were collected. The tissues were homogenized and sonicated on ice in sodium dodecyl sulfate (SDS) lysis buffer (Beyotime P00013C) with protease inhibitor cocktail (Roche Molecular Biochemicals) and phosphatase inhibitor (P2714, Sigma Aldrich), followed by centrifugation at 13,800g for 20 min at 4°C to obtain supernatant containing protein. Equal masses of protein from different supernatant were loaded and separated by SDS-polyacrylamide gel electrophoresis. After the transfer to a polyvinylidene difluoride membrane (Bio-Rad), blots were blocked and incubated at 4°C overnight with primary antibodies overnight at 4°C. The primary antibodies for Western blots in the present work include rabbit anti-p-GSK-3β (Ser9) (#9323, 1:1000, Cell Signaling Technology), rabbit anti-p-GSK-3β (Tyr216) (sc-135653, 1:1000, Santa Cruz), rabbit anti-β-catenin (#9582, 1:1000, Cell Signaling Technology), rabbit anti- GSK-3β (sc-9166, 1:1000, Santa Cruz), rabbit anti-BDNF (AB1534, 1:1000, Millipore), and mouse anti-β-actin (ab170325, 1:1000, Abcam). The blots were washed three times and incubated with horseradish peroxidase-conjugated IgG. The immune complex on the membrane was detected by enhanced chemiluminescence (Bio-Rad) and exposed by Tanon-5200 system. Western blot results were quantified using relative optical density (RelOD) by ImageJ software (National Institutes of Health, Bethesda, MD). These ratios were normalized to the control values.
Statistical analysis
All data were presented as means ± SEM. The results of behavioral tests were analyzed with repeated measures two-way analysis of variance (ANOVA) between groups and one-way ANOVA between testing days and treatment within group. The relative densities of Western blots and recognition indexes were compared using Student’s t-test. Statistical analysis was performed with SPSS 16.0. The value of p < 0.05 was considered significant.
Results
GSK-3β activity is decreased in spinal dorsal horn but increased in hippocampus following SNI
To evaluate the roles of GSK-3β in pain hypersensitivity and memory deficits, the levels of inactive p-GSK-3β (Ser 9) and active p-GSK-3β (Tyr 216) in spinal dorsal horn and in hippocampus were assessed at different time points after SNI, which produces neuropathic pain and STM deficits, persisting for at least 45 days (Figure 2). We found that GSK-3β activity was reduced in spinal dorsal horn but enhanced in hippocampus in SNI rats. Compared to sham rats, p-GSK-3β (Tyr 216) was downregulated and p-GSK-3β (Ser 9) was upregulated in spinal dorsal horn (Figure 1(a)), while in hippocampus, p-GSK-3β (Tyr 216) was upregulated and p-GSK-3β (Ser 9) was downregulated (Figure 1(b)). The opposite changes in GSK-3β activity started on day 3 after SNI and persisted for at least 45 days. The results suggest that SNI-induced pain hypersensitivity and memory deficits are associated with inhibition of GSK-3β in spinal dorsal horn and activation of the kinase in hippocampus, respectively.

The opposite changes in GSK-3β activity in spinal dorsal horn and in hippocampus produced by spared nerve injury. (a) and (b) Western blots show the changes in p-GSK-3β (Tyr216) and p-GSK-3β (Ser9) in spinal dorsal horn and in hippocampus 14 d after sham-operation (Sham) and at different time points after SNI. n = 4 in each time points, *p < 0.05, **p < 0.01, ***p < 0.001, compared with sham group (t-test). SNI: spared nerve injury.
Inhibition of GSK-3β prevents memory deficits produced by SNI but induces the behavioral signs of neuropathic pain in sham-operated animals
To determine the causal relationship between the opposite changes in GSK-3β activity in spinal dorsal horn and in hippocampus with the pain hypersensitivity and memory deficits induced by SNI, we tested the effects of GSK-3β inhibitor (SB216763) in SNI and sham rats. To do this, 23 rats were randomly divided into following four groups: vehicle (Vehi) + sham group (n = 5), Vehi + SNI group (n = 6), SB + sham group (n = 6), and SB +SNI group (n = 6). In the experiments, vehicle or SB216763 (5 mg/kg) was intraperitoneally injected in sham and SNI animals one day before and third and sixth days after surgeries. In line with the previous works,7,29 SNI produced mechanical allodynia and thermal hyperalgesia, manifested as decrease in PWTs and PWLs (Figure 2(a) and (b)) and reduced the recognition index of STM (Figure 2(c)), and the behavioral alterations persisted for at least 45 days.

Intraperitoneal injection of GSK-3β inhibitor Styrene-butadiene copolymer (SB) 216763 prevents STM deficits in SNI rats but induces persistent mechanical allodynia and thermal hyperalgesia in sham rats. (a) The experiment designs are shown. (b) and (c) The time courses of the changes in PWT and PWL in different groups as indicated are shown. Note that injection of SB216763 (Styrene-butadiene copolymer [SB], 5 mg/kg, i.p.) did not affect the decrease in PWT and PWL induced by SNI but induced mechanical allodynia and thermal hyperalgesia in sham rats. n = 5-6 in each group, ***p < 0.001, compared with Vehicle (Vehi) + Sham group (repeated measures two-way ANOVA). (d) In the same cohort of rats, SB216763 prevented the decrease in recognition index of short-term memory induced by SNI but had no effect on sham rats. **p < 0.01, compared with Vehi + Sham group. #p < 0.05, ###p < 0.001 vs. Vehi + SNI group (t-test). SNI: spared nerve injury; PWT: paw withdrawal threshold; PWL: paw withdrawal latency; NORT: novel object recognition test.
We found that pretreatment with SB216763 did not affect the behavioral signs of neuropathic pain in SNI rats, as PWTs and PWLs in SB + SNI group were not different from those in Vehi + SNI group (Figure 2(a) and (b)). Surprisingly, SB216763 injection decreased both PWTs and PWLs in sham rats, starting second injection, lasting for at least 45 days, indicating that inhibition of GSK-3β is sufficient to induce the behavioral signs of neuropathic pain. In the same cohort of rats, SB216763 prevented the decrease in recognition index of STM induced by SNI but did not affect the index in sham animals, as tested on days 7, 21, and 45 after surgeries (Figure 2(c)). To further evaluate the role of GSK-3β on memory deficits and neuropathic pain, we tested the effect of AR-A014418, another selective GSK-3β inhibitor 31 by intrathecal injection, as the compound cannot pass through blood–brain barrier (BBB). 32 The injection of AR-A014418 (400 ng/kg, 20 μl, daily) immediately before and one to six days after operations (Figure 3(a)) also did not affect the decrease in PWTs and PWLs in SNI rats but induced the behavioral signs of neuropathic pain in sham rats (Figure 3(a) to (c)). AR-A014418 also prevented the decrease in recognition index of STM induced by SNI, as tested 10 days after operation (Figure 3(d)). The results suggest that the drug injected intrathecally can also reach to brain. Consistently, we found that EB was detected in both spinal cord and brain, especially in hippocampus and the third ventricle, at 5 h after intrathecal injection (1%, 30 μl) (Figure 3(e)). The data indicate that inactivation of GSK-3β in spinal dorsal horn may contribute to neuropathic pain, while activation of GSK-3β in hippocampus contribute to memory deficits following SNI.

Intrathecal injection of GSK-3β inhibitor analytical reagent (AR)-A014418 prevents STM deficits in SNI rats but induces persistent mechanical allodynia and thermal hyperalgesia in sham rats. (a) The experiment designs are shown. (b) and (c) The injection of AR-A014418 (400 ng/kg, 20 μl) did not affect the decrease in PWT and PWL induced by SNI but induced mechanical allodynia and thermal hyperalgesia in sham rats. n = 5–6 in each group, ***p < 0.001, compared with Vehi + Sham group (repeated measures two-way ANOVA). (d) In the same cohort of rats, AR-A014418 prevented the decrease in recognition index of STM induced by SNI but had no effect on sham rats. **p < 0.01 compared with Vehi + Sham group, ###p < 0.001 vs. Vehi + SNI (t-test). (e) Representative photographs showing the Evans blue distribution (blue) in cerebrospinal fluid around spinal cord and brain (whole view, upper left), the ventral brain (bottom view, right), hippocampus, and the third ventricle (coronal view, left below) at 5 h after intrathecal injection (1%, 30 μl). SNI: spared nerve injury; BDNF: brain-derived neurotrophic factor; OD: optical density.
β-catenin and BDNF are upregulated in spinal dorsal horn but downregulated in hippocampus following SNI
How could the opposite changes of GSK-3β in spinal dorsal horn and in hippocampus lead to neuropathic pain and memory deficits following SNI? Previous works showed that the upregulation of β-catenin (a downstream molecule of GSK-3β) and BDNF in spinal dorsal horn is important for development of neuropathic pain, while the downregulation of them in hippocampus impairs memory.11,20–22 Accordingly, we hypothesized that the opposite changes in GSK-3β activity produced by SNI may differentially regulate β-catenin and BDNF expression in spinal dorsal horn and in hippocampus. To test this, we assessed the levels of β-catenin and BDNF in spinal dorsal horn and in hippocampus of SNI or sham rats treated with SB216763 or vehicle 10 days after surgeries. The results showed that in spinal dorsal horn (Figure 4(a)), the levels of both β-catenin and BDNF were significantly higher in Vehi + SNI group and in SB + Sham group, compared to Vehi + Sham rats. The data indicate that SNI upregulates β-catenin and BDNF in spinal dorsal horn, and inhibition of GSK-3β alone is sufficient to do so in sham rats. Compared to Vehi + SNI group, BDNF but not β-catenin was higher in SNI + SB group, that is, inhibition of GSK-3β may enhance BDNF but not β-catenin in spinal cord of SNI rats. In hippocampus (Figure 3(b)), however, the levels of both β-catenin and BDNF in Vehi + SNI group were significantly lower than those in Vehi + Sham group, indicating that SNI downregulates both β-catenin and BDNF. The downregulations of β-catenin and BDNF in hippocampus by SNI were completely abolished by injection of SB216763, as the levels of β-catenin and BDNF in SB + SNI group were significantly higher compared to vehicle + SNI group and were not different from those in vehicle + sham group. In sham rats, injection of SB216763 upregulated β-catenin but not BDNF. The results indicate that the decrease in GSK-3β activity in spinal dorsal horn is associated with the upregulations of β-catenin and BDNF, while the increase in GSK-3β activity in hippocampus with the downregulations of β-catenin and BDNF in SNI rats.

The differential effects of SNI and SB216763 injection on β-catenin and BDNF expression in spinal dorsal horn and in hippocampus. (a) and (b) The Western blots show the levels of β-catenin and BDNF in spinal dorsal horn and hippocampus in different groups as indicated. n = 3–4 in each group. **p < 0.01, ***p < 0.001 versus Vehi + Sham group (t-test), ##p < 0.01 versus Vehi + SNI group (t-test). STM: short-term memory; SNI: spared nerve injury; PWT: paw withdrawal threshold; PWL: paw withdrawal latency; NORT: novel object recognition test; EB: Evan blue.
Intravenous injection of IL-1β mimics the opposite molecular changes induced by SNI
Our previous work 13 show that IL-1β is increased in plasma following SNI, staring 5 h and persisting for at least 14 days with a peak as higher as 0.8 ng/ml on day 3 after surgery. Importantly, enhancing plasma IL-1β to 0.8 ng/ml in naïve rats by intravenous injection of recombination rat IL-1β (rrIL-1β, 100 ng/ml, 150 μl, three times per day) for successive seven days mimics the mechanical allodynia, STM deficits, and depressive behavior induced by SNI. Accordingly, we tested if intravenous injection of rrIL-1β in the same way may induce the opposite changes in GSK-3β, β-catenin and BDNF in spinal dorsal hornand hippocampus in naïve rats. Indeed, compared to vehicle-treated rats, in IL-1β-treated rats, inactive p-GSK-3β (Ser 9) was higher and active p-GSK-3β (Tyr 216) was lower in spinal dorsal horn, while in hippocampus, opposite changes were observed (Figure 5(a) and (b)). Both β-catenin and BDNF in spinal dorsal horn were higher but lower in hippocampus in IL-1β-treated rats than those in vehicle control rats (Figure 5(c) and (d)). The data suggest that the proinflammatory cytokine IL-1β is sufficient to induce the opposite molecular changes in the two regions.
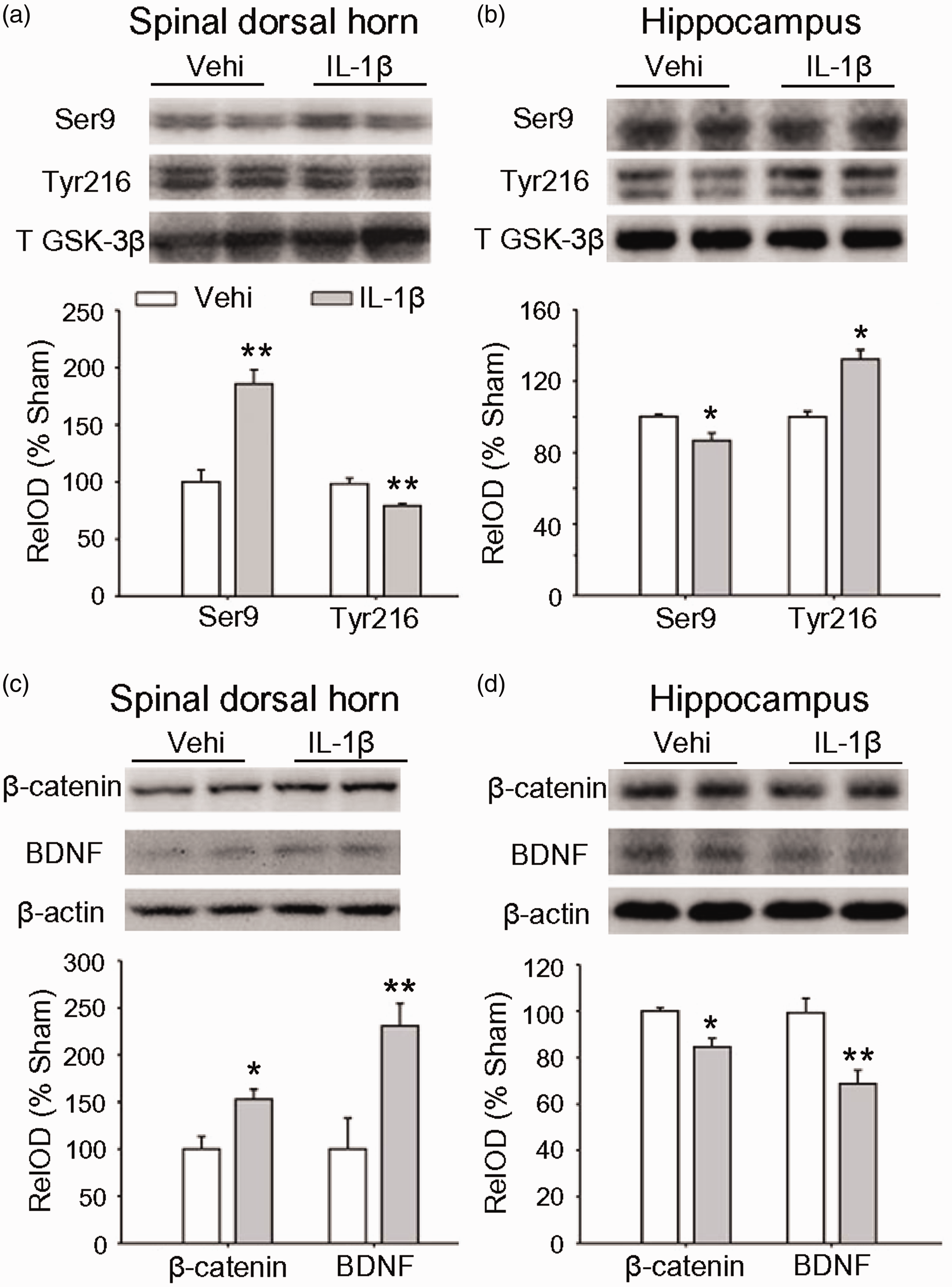
The opposite effects of intravenous rrIL-1β on the expressions of GSK-3β, β-catenin and BDNF in spinal dorsal horn and in hippocampus. (a) and (b) The levels of different forms of GSK-3β in spinal dorsal horn and in hippocampus after intravenous injection of Vehi or rrIL-1β (100 ng/ml, 150 μl, three times per day) for seven days are shown. (c) and (d) The repetitive injection of rrIL-1β upregulates β-catenin and BDNF in spinal dorsal horn and downregulated them in hippocampus. n = 3–4 in each group. *p < 0.05, **p < 0.01 versus Vehi group (t-test). GSK: glycogen synthase kinase-3beta; BDNF: brain-derived neurotrophic factor; OD: optical density.
Discussion
In the present work, we showed for first time that GSK-3β activity was reduced in spinal dorsal horn but enhanced in hippocampus in SNI model of neuropathic pain (Figure 1). Inhibition of GSK-3β by either intraperitoneal injection SB216763 or intrathecal injection of AR-A014418 prevented memory deficits induced by SNI but induced a chronic pain hypersensitivity in sham animals (Figure 2). Mechanistically, both β-catenin and BDNF expression were increased in spinal dorsal horn but decreased in hippocampus following SNI. SB216763 enhanced the upregulation of BDNF in spinal dorsal horn but prevented the downregulation of BDNF in hippocampus in SNI rats. In sham rats, injection of SB216763 led to the upregulations of β-catenin and BDNF in spinal dorsal horn, while in hippocampus, it affected neither of them (Figure 4). Finally, intravenous injection of IL-1β that induces mechanical allodynia, and memory deficits 13 mimicked the molecular changes in spinal dorsal horn and in hippocampus induced by SNI.
The role of GSK-3β on cognitive function and pain hypersensitivity
As mentioned in “Introduction” section, many preclinical studies show that GSK-3β inhibitors are effective for treatment of cognitive impairments caused by Alzheimer’s disease, Down syndrome, Parkinson’s disease, spinocerebellar ataxia, and traumatic brain injury (see King et al. 27 for a review). Inhibition of GSK-3β also potentiates the antidepressant-like effects of subthreshold doses of ketamine. 33 The present work further showed that inhibition of GSK-3β also prevented the SNI-induced memory deficits. There is no doubt that GSK-3β inhibitors are effective for treatment of cognitive disorders induced by different diseases or injuries. However, the present work revealed that inhibition of GSK-3β induced mechanical allodynia and thermal hyperalgesia in sham-operated rats, lasting for at least 45 days. The long-lasting side effect should be taken into count, when developing the drugs targeting GSK-3β.
Till date, the effect of GSK-3β inhibitor on pathological pain has been tested in only a few works. It has been shown that intraperitoneal, intrathecal, and subcutaneous injections of AR-A014418 inhibited pain responses induced by formalin, glutamate, and inflammatory cytokines in mice. 34 Later, the same group 35 showed that single intraperitoneal injection of GSK-3β inhibitor (AR-A014418) attenuated mechanical hyperalgesia induced by partial sciatic nerve ligation (PSNL) in mice, starting 30 min after injection and persisting for 2 h. AR-A014418 inhibited heat- and cold-hyperalgesia in PSNL mice but had no effect in sham mice. Another group 36 reported that the ratio of p-GSK-3β (Ser9)/total GSK-3β in spinal dorsal horn was decreased 3 days after PSNL but increased 10 days after the surgery, namely, GSK-3β activity was decreased in early stage and increased in late stage following PSNL. However, p-GSK-3β (Tyr216) was not assessed in this work. Intraperitoneal injection of AR-A014418 prior to PSNL and then daily attenuated mechanical and thermal hypersensitivity in PSNL rats six to eight days after application, and no effect was observed in sham rats. The two studies used the same model and same GSK-3β inhibitor with different animals (mouse vs rat), while the onset times for analgesic effect are quite different (30 min vs. six days). The reasons for the disagreement are unknown. However, our data showed that active p-GSK-3β (Tyr216) decreased and inactive p-GSK-3β (Ser9) increased in spinal dorsal horn following SNI, and the changes started 3 days and persisted for at least 45 days after surgery, indicating that SNI induces a long-lasting decrease in GSK-3β activity in spinal dorsal horn. Inhibition of GSK-3β by intraperitoneal injection of SB216763 or intrathecal injection of AR-A014418 did not affect mechanical allodynia and thermal hyperalgesia induced by SNI but caused the behavioral signs of neuropathic pain in sham rats. The results indicated that inhibition of GSK-3β in spinal dorsal horn may contribute to neuropathic pain induced by SNI. Both AR and SB suppressed GSK-3β activity by competing with ATP.31,37 Positron emission tomography studies show that SB21676337 but not AR-A01441831 is able to access into the central nervous system. Therefore, intraperitoneal injection of AR-A014418 may only act on peripheral nerve and tissues. This may partially explain the different results between our present and previous works.
The mechanisms by which GSK-3β improves cognitive function and induces neuropathic pain
In consistent with the clinical data that around two of three chronic pain patients suffer from memory deficits,2,3 our previous works showed that SNI induces neuropathic pain and impairs STM in rodents.6,7,11,13 However, the mechanisms underlying the comorbidity of chronic pain and memory deficits are still poorly understood. It is well established that the downregulation of β-catenin20,21 or BDNF38 in hippocampus impair memory, while the upregulation of β-catenin22,23 or BDNF11,16,17 in spinal dorsal horn is critical for the development of neuropathic pain. In the present work, we showed that β-catenin and BDNF were downregulated in hippocampus but upregulated in spinal dorsal horn following SNI (Figure 4). The opposite changes may contribute to the comorbidity of neuropathic pain and memory deficits after peripheral nerve injury.
Previous work show that the active GSK-3β degrades β-catenin and thus prevents its translocation to the nucleus for gene transcription.26,40 Consistently, our data showed that in SNI rats, the reduced GSK-3β activity was associated with enhanced β-catenin level in spinal dorsal horn, while in hippocampus, enhanced GSK-3β activity with reduced β-catenin. We found that injection of GSK-3β inhibitor SB216763 prevented the downregulations of β-catenin and BDNF in hippocampus induced by SNI. That is, prolonged increase in GSK-3β activity in hippocampus may lead to memory deficits via reducing β-catenin and BDNF. In spinal dorsal horn, injection of SB216763 upregulated both β-catenin and BDNF in sham rats, while enhanced the upregulation of BDNF but did not affect the upregulation of β-catenin in SNI rats. The data suggest that GSK-3β may also regulate BDNF by the factors other than β-catenin in spinal dorsal horn under pathological conditions. Further studies are needed to elucidate the mechanisms by which GSK-3β regulate β-catenin and BDNF expression in the two regions under physiological and pathological conditions.
The overproduction of IL-1β is sufficient to induce the opposite molecular changes in GSK-3β in spinal dorsal horn and in hippocampus
How can peripheral nerve injury differentially regulate GSK-3β, β-catenin, and BDNF in spinal dorsal horn and hippocampus? Our previous work 13 show that SNI induces mechanical allodynia and STM deficits and increases IL-1β in plasma, starting 5 h and persisting for at least 14 days after surgery, with a peak as high as 800 pg/ml. The behavioral changes are abolished by deletion of IL-1β receptor 1. Elevation of plasma IL-1β level to 1 ng/ml by intravenous injection of rrIL-1β (100 ng/ml, three times/day for seven days) mimics the effects of SNI. 13 The data suggest the increased plasma IL-1β is necessary and sufficient to induce mechanical allodynia and memory deficits in SNI rats. In this present work, we further showed that the same manipulation mimicked the opposite effects of SNI on GSK-3β, β-catenin, and BDNF in spinal dorsal horn and hippocampus induced by SNI (Figure 5). The data provide new insight for understanding the molecular mechanisms underlying the comorbidity of chronic pain and cognitive disorders mediated by abnormal expression of inflammatory cytokines. Further studies are needed to elucidate why peripheral nerve injury and the resultant IL-1β upregulation oppositely regulate GSK-3β in spinal dorsal horn and in hippocampus.
Previous studies have demonstrated that cytokines, including IL-1, tumor necrosis factor (TNF-α) in plasma are able to cross the BBB by saturable transport systems and directly affect the function of the central nervous system under physiological conditions (see Banks 41 for a review). In addition, either peripheral nerve injury or activation of primary afferent C-fibers also increases the permeability of BBB and blood–spinal cord barrier.42,43 Consistently, it has been shown that acute intravenous administration of rrIL-1β in rats induces Fos-like immunoreactivity in neurons of the paraventricular hypothalamic nucleus and elicits an activation of the pituitary-adrenal axis. 44 Our recent work shows that intravenous injection of rrIL-1β at a pathological concentration not only induces mechanical allodynia and memory deficit but also activates microglia in spinal dorsal horn and in many brain regions, including hippocampus. 13 Taken together, the data may explain the findings that enhancing plasma IL-1β level by peripheral nerve injury or intravenous injection of rrIL-1β induces opposite molecular changes in hippocampus and in spinal dorsal horn.
Besides produced by blood or peripheral immune cells, IL-1β is also synthesized and released by microglia, astrocyte, and even neurons in the brain and spinal cord.45–47 Moreover, IL-1 receptors are constitutively expressed at high levels in hippocampal neuron48,49 and sensory neurons.50,51 In hippocampus, IL-1β at physiological level promotes LTP and memory formation, 52 whereas at pathological high levels, IL-1β inhibit LTP, leading to memory deficits.53–56 In spinal dorsal horn, however, IL-1β facilitates synaptic transmission and induces spinal LTP57,58 and chronic pain. 59 These studies may explain why the increase of serum IL-1β results in chronic pain hypersensitivity and memory deficits. Similarly, overproduction of TNF-α in hippocampus and spinal cord also contribute to SNI-induced memory deficits and neuropathic pain by differentially regulating synaptic plasticity in the two regions.7,11,60
TNF-α that is constitutively expressed in nervous system is a leading cytokine in activating a cascade of other cytokines including IL-1β and IL-6.61,62 Many studies have unraveled that the synergistic interaction between TNF-α and IL-1β has profound effect on the proinflammatory process by forming the cytokine storm.63,64 Therefore, IL-1β and TNF-α may have similar actions in the development of chronic pain and cognitive impairment after peripheral nerve injury, and both of them are the potential targets for treating the disorders.
In summary, our data demonstrate that GSK-3β activity is reduced in spinal dorsal horn but enhanced in hippocampus following SNI. The opposite changes produced by upregulation of IL-1β contribute to chronic pain hypersensitivity and memory deficits by upregulation and downregulation of BDNF in the two regions. Therefore, GSK-3β inhibitors that improve memory may induce pain hypersensitivity.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by National Natural Science Foundation of China (No. 31771166, U1201223, 81771204, 81371198, and 81270377), Guangdong Training Plan for Excellent Young Teachers in Institutions of Higher Learning (No. Yq2013008), and Science and Technology Department of Guangdong (2015A020212020).