
Abstract
Persistent pain can occur after routine dental treatments in which the dental pulp is injured. To better understand pain chronicity after pulp injury, we assessed whether dental pulp injury in mice causes changes to the sensory nervous system associated with pathological pain. In some experiments, we compared findings after dental pulp injury to a model of orofacial neuropathic pain, in which the mental nerve is injured. After unilateral dental pulp injury, we observed increased expression of activating transcription factor 3 (ATF3) and neuropeptide Y (NPY) mRNA and decreased tachykinin precursor 1 gene expression, in the ipsilateral trigeminal ganglion. We also observed an ipsilateral increase in the number of trigeminal neurons expressing immunoreactivity for ATF3, a decrease in substance P (SP) immunoreactive cells, and no change in the number of cells labeled with IB4. Mice with dental pulp injury transiently exhibit hindpaw mechanical allodynia, out to 12 days, while mice with mental nerve injury have persistent hindpaw allodynia. Mice with dental pulp injury increased spontaneous consumption of a sucrose solution for 17 days while mental nerve injury mice did not. Finally, after dental pulp injury, an increase in expression of the glial markers Iba1 and glial fibrillary acidic protein occurs in the transition zone between nucleus caudalis and interpolaris, ipsilateral to the injury. Collectively these studies suggest that dental pulp injury is associated with significant neuroplasticity that could contribute to persistent pain after of dental pulp injury.
Introduction
Dental pain is one of the most prevalent forms of moderate to severe intensity acute pain experienced by adults and children. 1 A common source of dental pain is bacterially mediated infection of the dental pulp, the highly vascular and densely innervated soft tissue found within the tooth, which usually occurs as a consequence of tooth decay.2,3 The clinical diagnosis that defines this etiology is irreversible pulpitis, and it is often, but not always, extremely painful.4,5 As the infection progresses throughout the pulpal tissue, the pulp degenerates and ultimately becomes completely necrotic, while the inflammation continues to spread in the periodontal tissues surrounding the tooth. As part of this degenerative process, damage and die-back of the trigeminal afferents innervating dental pulp occurs. Root canal treatment is commonly performed to relieve pain by removing inflamed or necrotic pulpal tissues and eliminating the bacterial infection inside of the tooth structure. 6 Recent researches on outcomes after root canal treatment have determined that between 3% and 12% of patients will have chronic pain after seemingly successful root canal treatment.7–9
Studies using animal models of injury or disease that produce hypersensitivity and/or spontaneous pain have identified multiple mechanisms associated with chronic pain. Inflammatory mediators released in the injured tissues activate sensory afferents, producing peripheral and central sensitization. 10 With the development of central sensitization, there is modulation of central synaptic connectivity that causes amplification of nociceptive pain signaling and opens a pathway for non-nociceptive stimuli to cause pain. 11 Upregulation and activation of microglia and astrocytes contribute to the neuroplasticity within the central nervous system (CNS), which appears to be associated with developing persistent pain.12,13 Further, damage to primary afferents produces changes in the gene expression in the damaged and adjacent neurons, which could contribute to the development of persistent pain states.14,15 The development of clinical neuropathic pain, a type of chronic pain in which a lesion or disease affecting the somatosensory system can be identified, highlights the importance of nerve damage as a route of pain chronification. 16 Nerve injury animal models of neuropathic pain have identified markers including neuropeptide Y (NPY) and activating transcription factor 3 (ATF3), that appear to be specifically associated with peripheral nerve injury.17,18 Persistent mechanical allodynia is another common feature of nerve injury and chronic inflammation in animal models.19–21 Although it is unclear which of these processes are essential to the development of chronic pain state, they can be thought of as indicators of potentially irreversible pathological processes occurring within the somatosensory system.
In this study, we identified that some neurobiological mechanisms that contribute to the development of persistent pain are in fact activated in a mouse model of pulp injury. The clinical implications for the engagement of these pathological processes in the trigeminal system after pulpal injury are important. They include the potential for pain chronicity due to damage sustained by primary afferent fibers after bacterial-mediated pulpal inflammation and degeneration, or iatrogenic surgical removal of pulp tissues during root canal treatment.
Materials and methods
Animals
All experiments were approved by the Institutional Animal Care and Use Committee at the New York University College of Dentistry and followed the guidelines provided by the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Adult female C57BL/6 and Balb/c mice were purchased from Charles River Laboratory. The mice had freely available food (Picolab© Rodent Diet 20, pellets) and water and were maintained in a facility with a 12-h light/dark cycle and housed in groups of 3 to 5. Experimental manipulations were performed at 10 to 12 weeks of age.
Dental pulp injury
To produce inflammation and degeneration of the dental pulp, a mechanical exposure of the pulp tissue was made as previously described. 22 Briefly, mice (18–25 g) were anesthetized by intraperitoneal (i.p.) injection of ketamine-xylazine anesthesia, 100 mg/kg and 10 mg/kg, respectively. The mouth of the mouse was propped open, and the enamel and dentin of the left first and second maxillary molars were drilled with a ¼ round dental bur at low speed until the inner chamber containing pulpal tissue was exposed (Supplementary Figure 1). The pulp exposures were visually confirmed using an illuminated operating microscope. Sham mice received the same anesthesia and had their mouths held open with forceps for a comparable time.
Mental nerve injury
An adapted version of the mental nerve ligation surgery was used to model orofacial neuropathic pain, by producing a chronic injury to the mental nerve, a terminal branch of the mandibular nerve innervating the lower lip. 23 Briefly, mice were anesthetized with a combination of i.p. ketamine-xylazine anesthesia, 100 mg/kg and 10 mg/kg, and the mental nerve was bilaterally exposed at its point of exit from the mental foramen, in the anterior mandible. The left and right mental nerves were individually ligated using Ethilon silk (4.0) suture material. Controls included naïve controls, which received anesthesia only, nerve transection, in which the mental nerve was completely cut, and sham-operated mice, which received the same anesthesia and bilateral exposure of the mental nerves without ligation.
Measurement of mechanical thresholds
Mice (Balb/c females) were exposed to the experimenter, testing room, and chambers during multiple sessions over a one-week acclimation period before baseline testing began. Balb/c mice were used because we attempted to measure orofacial mechanical thresholds, and this strain of mice was more amenable to the handling and restraint required for this test, although we ultimately found the orofacial mechanical testing unreliable. Animals were also acclimated to their surroundings for 30 min prior to each testing session. Three baseline measurements were made prior to injury, on three different days, and mice were tested at multiple time points after dental pulp injury (DPI) or mental nerve injury (MNI). During the testing procedure, mice were contained in a transparent plexiglass container placed over wire mesh surface, and the left and right hindpaws were stimulated with standardized von Frey filaments by a blinded experimenter using the up-and-down method. 24
Sucrose consumption assay
Mice (C57BL/6 females) were evaluated for the amount of sucrose solution they would voluntarily consume as previously described. 22 The C57BL/6 strain was used because the sucrose consumption assay was validated in this strain. Food and water were removed from the home cages 3.5 h before the start of the experiment. At the time of food removal, the mice were weighed, and the cages were brought into the testing room. The sucrose consumption experiment was initiated 40 min before the start of the dark cycle. The test began when each animal was individually placed in the test cage containing one bottle filled with a 2.5% to 5% sucrose solution. The animals would drink freely for 2 h, after which they were removed from the test chambers and returned to their home cages where they were group housed. Sucrose bottles were weighed before and after the test.
Mice were initially trained in the sucrose consumption assay using a 5% sucrose on two separate test days. Mice were further trained with a 2.5% sucrose solution for three to four additional days, to establish baseline consumption. Test days were non consecutive, and typically three tests per week were performed. Mice were assigned to control or injury groups, in a manner allowing for a stratified distribution of baseline levels of sucrose consumed between treatment groups, and received DPI, MNI, or sham procedures. At 21 days post-injury, mice were randomly assigned to receive either a gabapentin (10 mg/kg) or saline i.p. injection. Mice were briefly lightly anesthetized with isoflurane anesthesia (1.5%) prior to the injection. Mice were acclimated to the injection procedure for three days leading up the drug experiment, by performing brief isoflurane anesthesia, and handling to simulate injection procedures. The drugs were administered 4 h prior to the sucrose consumption test.
Immunohistochemistry and histology
Immunohistochemistry (IHC) and jaw histology were performed as previously described in literature. 25 Briefly, female C57BL/6 J mice were sacrificed at various time points post-injury by overdose with 2.5% avertin, then perfused transcardially with 10 mL of 0.1 M phosphate-buffered saline (PBS) and 30 mL of 10% formalin in 0.1 M phosphate buffer (PB). The trigeminal ganglia (TG), brainstem, and maxillary jaws were dissected and post-fixed for 30 min (TGs) or 4 h (brainstems and jaws) at 4℃ and then cryoprotected overnight in 0.1 M PB with 30% sucrose. Tissues were embedded in optimal cutting temperature compound (Sakura Finetek USA, Torrance, CA, USA) and stored at −80℃ until cryosectioning. Brainstem, TG, and maxilla tissues were sectioned at 40 µm, 16 µm, and 40 µm, respectively. Sections of TG and maxilla were mounted onto Superfrost Plus Microscope Slides (Fisher Scientific, Pittsburgh, PA, USA) and kept frozen at −80℃ until use, while brainstem sections were collected as floating sections.
For IHC, sections of TG or maxilla were blocked for 60 min in 5% normal goat serum in 0.1 M PB with 0.3% Triton X-100 normal goat serum (NGST), while brainstem sections were blocked with 0.5% Triton-X and 0.5% bovine serum albumin in 1X PBS. Sections were incubated overnight with primary antibody diluted in 5% NGST. The following day sections were washed 3X with 1% NGST, then incubated for 2 h with secondary antibody (Alexa-488, Alexa-594, Streptavidin 594, Life Technologies Corporation, Grand Island, NY, USA, diluted 1:700 in NGST). After washing 3X in PBS, sections were mounted, briefly air dried, and coverslipped with Aqua-mount. Primary antisera and lectins used for staining included the following: rabbit anti-ATF3 (sc-188, 1:800, Santa Cruz Biotechnology, Dallas, TX, USA), guinea pig anti-Substance P (1:1000, ab 10353, Abcam, Cambridge, MA, USA), biotinylated isolectin B4 (IB4, Vector Laboratories Inc., Burlingame, CA, USA, 1.5 h), mouse anti-N52 (N0142, 1:10,000, Sigma, Saint Louis, MO, USA), rabbit anti-glial fibrillary acidic protein (GFAP, Abcam ab7260, 1:1000), and rabbit anti-Iba1 (Wako,019-19741, 1:500).
To quantify cell labeling in the TG, two to three images were captured from five to seven non-consecutive sections, per animal, from the region of the ganglion innervating the injury location, i.e., the maxillary region for DPI mice and mandibular region for MNI mice. Images were captured using a Nikon Eclipse Ti microscope. The number of cells expressing specific immunoreactivity were counted, and the percentage of immuno-positive cells per section was averaged for each animal.
Glial immunoreactivity was quantified from non-consecutive brainstem sections from the nucleus caudalis, and the transition zone between caudalis and interpolaris. 26 We focused our analysis on the outer lamina in the dorsolateral regions of these nuclei, as we (unpublished), and others have previously observed induction of c-fos expression in response to dental pulp stimulation in these regions. 27 Anatomical studies have also confirmed these regions receive projections of pulpal afferents. 28 In captured images, 1,100 µm2 region of interest was selected bilaterally from immunostained sections. The mean intensity of fluorescence was measured from each region of interest, and background fluorescence intensity was subtracted. Acquisition settings were optimized to the immuno-target (GFAP or Iba1), and the settings were maintained for the duration of capturing all images.
Morphological changes in the dental pulp of mice with DPI were examined by a comparison of injured and uninjured tissues with hematoxylin and eosin staining. Tissues selected for hematoxylin and eosin staining were stained with hematoxylin solution and differentiated in 1% acid alcohol. After sufficient staining, sections were transferred to 0.2% ammonia water for bluing and then into eosin solution with washes in deionized water between steps.
Quantitative polymerase chain reaction
Quantitative polymerase chain reaction (PCR) was performed to evaluate transcript levels of two genes associated with nerve injury, NPY, and ATF3, and two genes associated with inflammatory signaling, tachykinin precursor 1 (TAC-1) and CALCA (calcitonin gene-related peptide), at 3, 21, or 42 days following DPI and 5 or 42 days following MNI. Briefly, TGs were extracted, snap-frozen, and stored at −80℃ until RNA isolation. RNA was extracted by crushing frozen TG in a 1.5-mL tube using a pestle, followed by addition of TRI reagent and further following the manufacturer’s instructions for extraction. Concentration was determined using a NanoDrop spectrophotometer. RNA samples were diluted to a concentration of 85 ng/mL. Reverse transcription was performed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to manufacturer’s instructions. Real-time (RT) PCR was performed using EXPRESS qPCR SuperMix Universal (Invitrogen) and Taqman Gene Expression Assays (Mouse ATF3, Cat# Mm00476032_m1; TAC-1, Mm01166996_m1; NPY, Mm01410146_m1; CALCA, Cat# Mm008014, GAPDH, Mm99999915_g1, Life Technologies) for each gene. Each reaction was performed at least in duplicate, and for each gene, reactions containing no template and non-reverse-transcribed RNA were included, as controls.
Statistical analysis of data
Results are expressed as the means ± SEM of values obtained from individual mice. Statistical analysis and graphical representation of data were performed with Graph Pad Prism 5 software (Graph Pad Software Inc., La Jolla, CA, USA). Time-course effects were analyzed for statistical significance by a two-way analysis of variance (ANOVA) followed by a Bonferroni’s test for multiple comparisons. A one-way ANOVA followed by a post hoc Dunnett’s test was used for multiple comparisons with the same control group or by a post hoc Bonferroni’s test for comparison between groups. Only probability values (p) less than 0.05 were considered statistically significant.
Results
Effect of DPI on mRNA levels of ATF3, NPY, TAC-1, and CALCA
Mechanically exposing the dental pulp of a molar tooth causes a progressive degeneration of this densely innervated and highly vascularized soft tissue, housed inside the tooth (Supplementary Figure 1(a) to (d)). As the pulpal tissue adjacent to the exposure succumbs to bacteria, it becomes necrotic, and the primary afferent fibers within the pulp retreat or die-back. As such, there is significant inflammation and remodeling of the peripheral afferents in this type of injury (Supplementary Figure 1(b)).
In order to test the hypothesis that genes associated with nerve injury are upregulated
in chronic pulpitis, we measured mRNA expression of ATF3 and NPY in the TG at various time
points after pulp injury. ATF3 is a transcription factor and a sensitive marker of injured
primary afferent neurons.
17
The mean level of ATF3 mRNA expression in the TG ipsilateral to the
pulp injury was slightly, but significantly, increased relative to the contralateral TG,
and the time after injury did not affect expression levels (Figure 1(a); two-way repeated measures (RM) ANOVA:
Side: p = 0.005). Post hoc analyses did not identify differences at
individual time points, and expression was higher on the ipsilateral side at all time
points after pulp exposure (3 days: 32% increase ipsi vs. 19% increase contra, 21 days:
21% ipsi vs. 6% increase contra, and 42 days: 34% ipsi vs. 7% increase contra; percent
change relative to shams). The expression of NPY is increased in a subset of primary
afferent fibers after nerve injury, and endogenous NPY functions as an inhibitor of
hyperalgesia.18,29,30 Similar to ATF3, the mean level of NPY
mRNA in the TG ipsilateral to injury was slightly higher relative to the contralateral TG
at all time points (3 days: 366% increase ipsi vs. 240% increase contra; 21 days: 8%
decrease ipsi vs. 20% decrease contra; 42 days: 99% increase ipsi vs. 35% increase contra;
percent change relative to shams) (Figure
2(b); two-way RM ANOVA: Side: p = 0.09; Bonferroni post hoc
significant on Day 3 (p < 0.05)). Interestingly, increased expression
of genes associated with inflammatory signaling pathways was not observed. In fact,
expression of the TAC-1 gene, which encodes the tachykinins substance P (SP) and
neurokinin A, both important mediators of painful inflammatory responses, was lower in the
ipsilateral versus the contralateral TG at every time point (3 days: 9% vs. 44% increase;
21 days: 12% decrease vs. 12% increase; 42 days: 39% vs. 65% increase, relative to shams;
Figure 2(c); two-way RM ANOVA:
Side: p = 0.08). Finally, there was no effect of injury on transcript
levels of the CALCA gene, which encodes the pro-inflammatory neuropeptide calcitonin
gene-related peptide, in the ipsilateral versus contralateral TG (Figure 2(d); two-way RM ANOVA: Side:
p = 0.7). In sum, ATF3 and NPY gene expression, both associated with
nerve injury, were more highly expressed in the TG ipsilateral to pulp injury, while such
increases were not observed for the pro-inflammatory genes TAC-1 and CALCA. RT-PCR analysis of mRNA levels of ATF3, NPY, TAC-1, and CALCA in trigeminal ganglia
after unilateral dental pulp injury or mental nerve injury. Transcript levels were
normalized to GAPDH levels in each sample. Fold change was calculated relative to
sham mice in DPI animals and to naïve mice in MNI animals. For DPI, mice analyses
compared the levels expressed in the TG ipsilateral to the pulp exposure relative to
the contralateral TG (a) to (e). (a) ATF3 mRNA levels were significantly higher in
the ipsilateral TG versus the contralateral TG (two-way RM ANOVA, Side:
p = 0.005, F = 9.8, DOF = 1;
Time: p = 0.75, F = 0.3, DOF = 2;
Interaction: p = 0.63, F = 0.5,
DOF = 2). Bonferroni post hoc non-significant (b): NPY mRNA
levels were higher at all time points in the ipsilateral TG and were marginally
statistically significant (two-way RM ANOVA: Side: p = 0.09,
F = 3.1, DOF = 1; Time:
p = 0.4, F = 0.96, DOF = 2;
Interaction: p = 0.5, F = 0.8,
DOF = 2). Bonferroni post hoc analysis significant on Day
3 p < 0.05. (c) TAC-1 mRNA levels were lower at all times
ipsilateral to injury, and this effect was marginally significant (two-way RM ANOVA:
Side: p = 0.08, F = 3.4, DOF = 1;
Time: p = 0.5, F = 0.7, DOF = 2;
Interaction: p = 0.5, F = 0.9,
DOF = 2). (d) CALCA mRNA did not differ by side of injury over
time (two-way RM ANOVA: Side: p = 0.7; Time:
p = 0.7; Interaction: p = 0.1). (e) There was no
difference in ATF3 mRNA levels in TGs of mice with mental nerve ligation (MNI) or
transection relative to sham or naïve mice (two-way ANOVA: Side:
p = 0.8; Time: p = 0.1; Interaction:
p = 0.5). N = 5 to 13 animals per time
point. After DPI or MNI, ATF3 (green (a) to (d)) is more often expressed in the cells of
the trigeminal ganglia. (a) ATF3 expression is induced in the nuclei of trigeminal
ganglion cells ipsilateral to DPI 21 days after injury. (b) ATF3 was not induced in
trigeminal ganglion cells contralateral to DPI. (c) ATF3 was induced in trigeminal
ganglia neurons after MNI at 21 days after injury. (d) ATF3 was not induced in
trigeminal ganglia neurons at 21 days after sham MNI surgery. Scale
bar = 100 µm.

To give some context to the magnitude of the observed effects of pulp injury on gene expression, we performed similar analyses in mice with a frank nerve injury. To accomplish this, we utilized a rodent model of orofacial neuropathic pain in which the mental nerves are ligated.31–33 We chose the mental nerve, as it is a terminal branch of the mandibular nerve that innervates a relatively constrained anatomical area, so that the magnitude of the injury to primary afferents might be comparable to that created by a pulp injury in two molar teeth. Surprisingly, we found no effect of MNI on ATF3 gene expression, even though multiple time points were evaluated (Figure 1(e)). Possible reasons for this are expanded on in the discussion, but our interpretation is that it is difficult to detect changes in ATF3 expression in whole TG preparations after injury to small, terminal nerve branches, like the mental nerve. Given this, the small but consistent increase in ATF3 mRNA observed after DPI, is more likely to be physiologically meaningful. To further investigate neuroplasticity after pulp injury, we next quantified cellular expression of neuronal markers using IHC.
Effect of DPI and MNI on ATF3, SP, and IB4 labeling in TG
To determine whether the number of cells expressing immunoreactivity for ATF3 and SP, and
IB4 binding, is affected by pulp injury, we used IHC to quantify the proportion of TG
neurons expressing these markers after unilateral DPI (Figures 2 and 3). Similar to the RT-PCR findings, we found a slight
but significant increase in the number of neurons expressing ATF3 in the TG ipsilateral to
the DPI, relative to the contralateral TG (Figures 2(a) and (b) and 4(a): two-way RM ANOVA: Side:
p = 0.006; mean percent expression assessed out of 5914 cells on average
per animal). The increase in cells expressing ATF3 was most pronounced between 7 and 28
days after injury, with the largest increase observed at 28 days (a 44% increase vs. the
contralateral side; Figure 4(d)).
We also assessed expression of two other markers of nociceptors, SP, a pro-inflammatory
neuropeptide increased in primary afferents innervating inflamed tissues and decreased
after axotomy or ligation (although upregulation in myelinated afferents has been
reported),34,35 and IB4, a cell surface
marker of unmyelinated, mostly non-peptidergic neurons, which is decreased in axotomized
primary afferents.
36
Interestingly, we found that SP levels were slightly lower in the ipsilateral TG at every
time point except for the three-day time point (Figures 3(a) and (b) and 4(c) and (f); two-way RM ANOVA: Side:
p = 0.05, no effect of time). These findings parallel the RT-PCR results,
for the TAC-1 gene, which encodes SP (Figure 1(c)). Pulp injury did not affect the number of neurons binding IB4
(Figures 3(c) and (d) and 4(b) and (e); p = 0.9). After DPI, SP (red (a) and (b)) is expressed in slightly fewer neuronal cells in
the trigeminal ganglion, while the number of cells labeled with IB4 (red (c) and
(d)) is unchanged. A comparison was made between immunoreactivity in the trigeminal
ganglion ipsilateral to the injury ((a) and (c)) relative to the contralateral TG in
the same mouse ((b) and (d)). Effect of unilateral DPI or MNI on the percentage of neurons immunoreactive for
ATF3 and SP, and labeled with IB4 in trigeminal ganglion, assessed by IHC. Cells
were quantified at various time points after DPI (a) to (f) and at 21 days after MNI
(g) to (i). Data are presented as the percentage of total counted TG cells
immunoreactive for each marker. Averages show the mean % of labeled cells counted in
each mouse (DPI n = 3–4; MNI n = 3–5). (a) ATF3 is
expressed in more cells in the TG ipsilateral to pulp injury (two-way RM ANOVA:
Side: p = 0.007, F = 9.3,
DOF = 1; Time: p = 0.8, F = 0.5,
DOF = 5; Interaction: p = 0.8,
F = 0.5, DOF = 5). (b) IB4 staining did not
differ in the ipsilateral TG relative to the contralateral after pulp injury
(two-way RM ANOVA: Side: p = 0.9; Time: p = 0.5;
Interaction: p = 0.4). (c) SP is expressed in fewer cells in the TG
ipsilateral to pulp injury relative to the contralateral TG (Side:
p = 0.05, F = 4.6, DOF = 1;
Time: p = 0.9, F = 0.2, DOF = 5;
Interaction: p = 0.1, F = 1.9,
DOF = 5). (d) to (f) Percent change in the immunoreactivity of
the markers ATF3, and SP, and labeled with IB4 in ipsilateral versus contralateral
TG. Each data point is the change observed in an individual mouse (g) A greater
percentage of TG cells express ATF3 in mice with MNI relative to mice with sham
surgery and naïve mice (one-way ANOVA, p = 0.003,
F = 12.8, DOF = 2; post hoc Dunnett’s Multiple
Comparison Test NI vs. Naïve p < 0.01 NI vs. Sham
p < 0.01). (h) There was no effect of MNI on the percentage of
TG cells labeled with IB4 (one-way ANOVA, p = 0.4). (i) There was
no effect of MNI of the percentage of TG cells expressing SP (one-way ANOVA,
p = 0.9).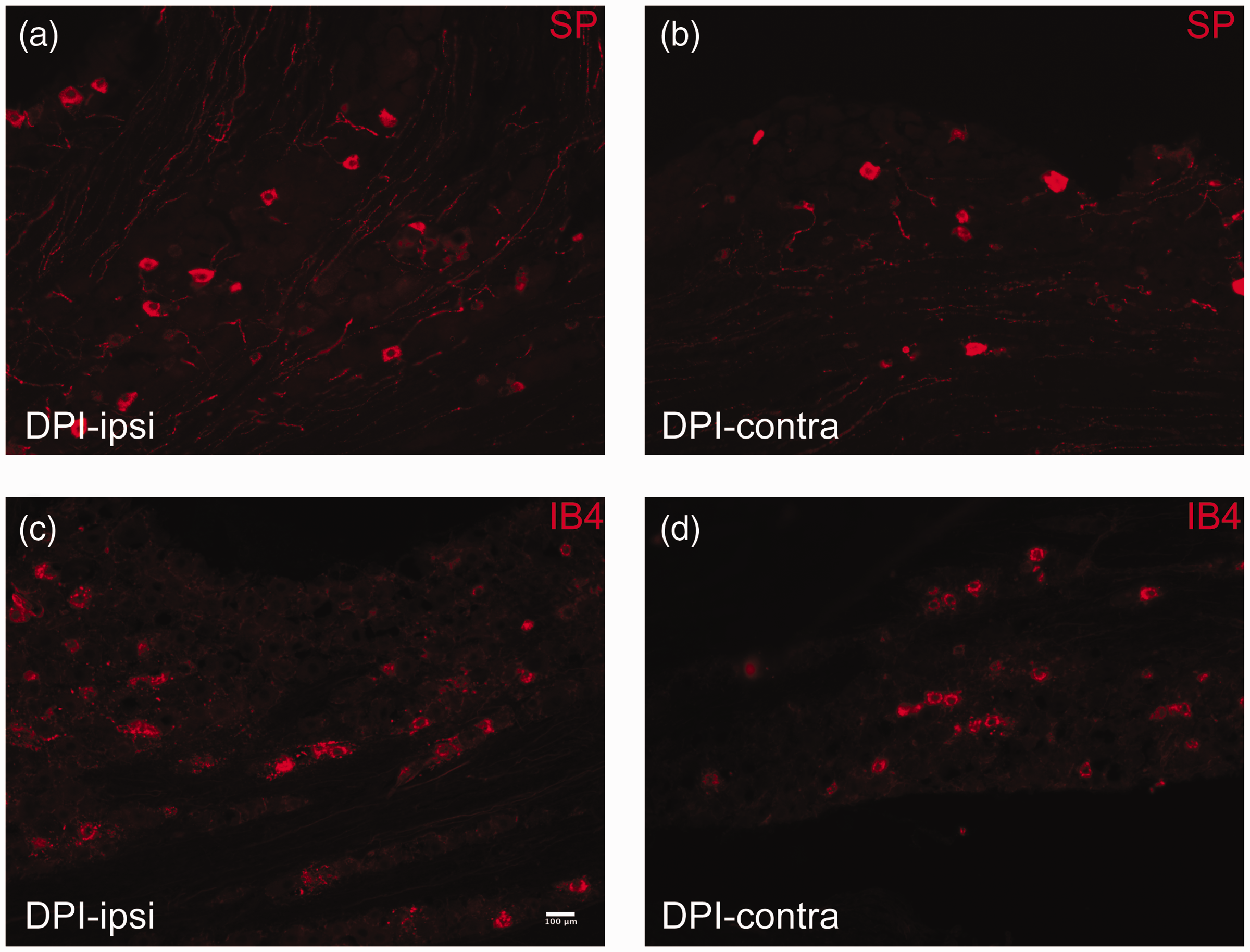

For comparison, we determined whether bilateral ligation of the mental nerve produced a change in ATF3 expression, 21 days after surgery. In contrast to the RT-PCR findings, where we saw no effect of nerve injury on ATF3 mRNA expression, we found a significant increase in the number of TG neurons expressing ATF3 in animals with MNI relative to sham and naïve animals (Figure 4(d): one-way ANOVA; p = 0.003, mean percent expression assessed out of 1173 cells (avg) per animal). There was no effect of nerve injury on the percentage of TG neurons labeled with IB4 (Figure 4(h), p = 0.1) or immunoreactive to SP (Figure 4(i), p = 0.5).
Effect of DPI and MNI on hindpaw mechanical hypersensitivity
We further compared several behavioral and physiological outcomes in mice receiving DPI,
MNI, or sham treatments. We attempted to measure withdrawal thresholds directly in the
face in freely moving, but contained Balb/c mice, but found that the results were
unreliable due to high variability. Instead paw withdrawal thresholds were used, as rodent
models of headache produce a reduction of mechanical withdrawal thresholds in the hindpaw,
which is reversible with analgesics.37,38 We found that DPI and MNI cause a
significant reduction in mechanical withdrawal thresholds in the hindpaw relative to
controls (Figure 5(a), DPI two-way
RM ANOVA: Treatment: p = 0.002, Time: 0.0007, Interaction: 0.002; Figure 5(b), MNI two-way ANOVA:
Treatment: p < 0.0001, Time: 0.005, Interaction:
p < 0.0001), but that the effects of DPI were transient, returning to
baseline by 12 days after injury, while mice with MNI still had mechanical allodynia in
the hindpaw at the last evaluated time point, 36 days after injury. It is unclear whether
the change in mechanical hypersensitivity in the hindpaw has any relation to ongoing
spontaneous pain. Both bilateral DPI and MNI produce hypersensitivity to cutaneous mechanical
stimulation in the hindpaw. (a) Withdrawal thresholds to mechanical stimuli applied
to the hindpaw were measured before and after DPI and a highly significant effect of
the factors analyzed was found (two-way RM ANOVA: Injury:
p = 0.002, F = 19.6, DOF = 1;
Time: p = 0.0007, F = 8.0,
DOF = 3; Interaction: p = 0.002,
F = 6.8, DOF = 3). Bonferroni
multiple-comparison test was used for post hoc analysis, demonstrating a significant
difference at Days 2 and 6 after surgery, but no difference between groups at Day
12. N = 5 per group. (b) Withdrawal thresholds to mechanical
stimuli applied to the hindpaw were measured before and after MNI. Two-way ANOVA
showed a significant effect of the factors analyzed (Injury:
p < 0.001, F = 137.6, DOF = 1;
Time: p = 0.005, F = 3.6,
DOF = 5; Interaction: p = <0.0001,
F = 7.0, DOF = 5). Bonferroni post hoc analysis
showed significant differences between the two groups at all tested time points.
N = 6 to14 per group at different time points (combination of two
experiments).
Effect of DPI and MNI on voluntary consumption of sucrose solution and change in body weight
Previous work established that after DPI, mice spontaneously consume more of a 2.5%
sucrose solution than controls during a 2-h test. Mice with DPI also gained less weight
per day in the 48 h after surgery than control mice, despite comparable food consumption
levels.
22
We
performed an experiment using mice with bilateral DPI, DPI sham, bilateral MNI, or MNI
sham to determine if both types of orofacial injury models affect spontaneous sucrose
consumption (Figure 6). First, as
previously reported, we observed that DPI mice consume more sucrose than sham DPI mice and
these changes persisted out to at least 17 days (Figure 6(a) mean group averages, two-way RM ANOVA:
DPI: p = 0.04; Figure
6(c) mean of individual changes in consumption from baseline, two-way RM ANOVA:
Treatment group: p = 0.02) Interestingly, mice with MNI did not consume
more sucrose than sham MNI mice, in fact the trend was for them to drink less (Figure 6(b), two-way RM ANOVA: Injury:
p = 0.5). Comparing the average amount of sucrose solution consumed
during each test after injury, mice with MNI, MNI sham, and DPI sham all drank
significantly less after injury than mice with DPI (Figure 6(d), one-way ANOVA,
p = 0.02). Thus, increased sucrose consumption occurs after DPI but not
after MNI. Effect of DPI or MNI on consumption of a 2.5% sucrose solution. Mice were
acclimated to the assay and the amount of solution spontaneously consumed during a
2-h test period was measured before and after injury. (a) Group means at baseline
(BL) and multiple time points after injury in DPI and DPI sham animals. Animals in
the DPI group consumed significantly more sucrose than shams (two-way RM ANOVA:
Injury: p = 0.04, F = 6.1,
DOF = 1; Time: p = 0.10, (ns); Interaction:
p = 0.06 (ns)). Bonferroni post hoc analysis showed a significant
increase on Day 8, p < 0.05. (b) Group means at BL and multiple
time points after injury in MNI and MNI sham animals. There was no effect of MNI on
sucrose consumed (two-way RM ANOVA: Injury: p = 0.5; Time:
p = 0.8; Interaction: p = 0.6). (c) Group means
of each mouse subjects’ change in sucrose consumption from BL at multiple time
points after injury. There was a significant effect of treatment group on sucrose
consumption (two-way RM ANOVA: Treatment group: p = 0.04,
F = 4.1, DOF = 3; Time: p = 0.3
(ns); Interaction: p = 0.8 (ns)). Bonferroni multiple comparisons
post hoc test showed a significant difference between DPI and DPI sham on Day 8
after injury (p < 0.05). (d) Overall group differences showed
that the increase in sucrose consumption after injury only occurred in the DPI mice.
The average amount of sucrose consumed by mouse at every test date after injury was
calculated and the means of the treatment groups compared (one-way ANOVA,
p = 0.02, F = 4.3, DOF = 3)
Newman-Keuls Multiple Comparison post hoc test showed the DPI group to be
significantly different than each of the other treatment groups
(p < 0.05). N = 5 mice/group for all data.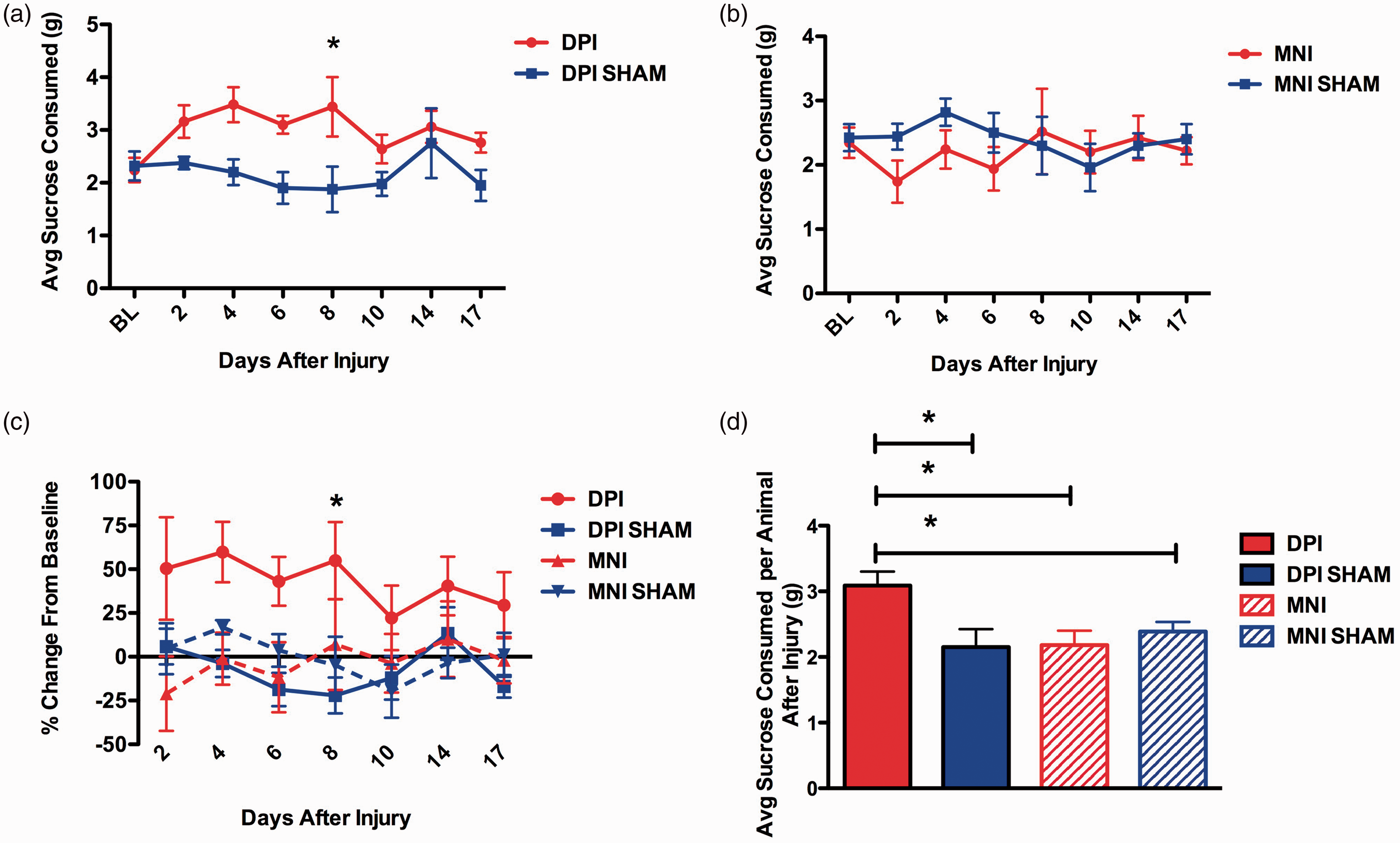
We attempted to test whether administration of gabapentin, an analgesic used clinically to treat neuropathic pain, would reverse the increase in sucrose consumption observed in DPI mice. We observed that the mice with DPI decreased their sucrose consumption whether given active drug or vehicle, so we were unable to conclude what the effect of gabapentin was on DPI-evoked increase in sucrose consumption (Supplementary Figure 2). Finally, we determined the effects of DPI and MNI on weight changes after injury. As previously reported, we found that DPI decreased the amount of weight gained in the first four days after surgery (Supplementary Figure 3(a) and (c)). However, no difference was observed between MNI and MNI sham animals (Supplementary Figure 3(b) and (c)).
Effect of DPI on the expression on glial markers in the trigeminal nucleus
Increased expression, activation, and inflammatory signaling in microglia and astrocytes
of the CNS is an important mechanism for the maintenance of pathological pain, including
neuropathic pain, and has been demonstrated in many preclinical pain models.12,39 We examined the effect of DPI on the
expression of GFAP, a marker of astrocytes, and Iba1, a marker of microglia, in regions of
the trigeminal nucleus known to receive projections from pulpal afferents and to be
important for pain processing.40,41 Five
days after unilateral DPI, we found increased GFAP immunoreactivity on the injured side
relative to the contralateral side, in both the nucleus caudalis and transition zone of
the trigeminal nucleus (Figures
7(a) and (b) and 8(a)).
Although smaller in magnitude, we also found a significant increase in Iba1 expression on
the injured side in the transition zone of the trigeminal nucleus (Figures 7(c) and (d) and 8(b)). Effect of unilateral DPI on expression of glial markers in the nucleus caudalis and
transition zone of the caudalis and interpolaris. Mice underwent procedures to
expose the dental pulp of the first and second maxillary molars on one side. Seven
days later, mice were euthanized and brainstems collected and processed for IHC. (a)
GFAP immunofluorescence (green) ipsilateral to DPI in the transition zone, (b) Less
GFAP immunofluorescence is observed on the contralateral size. (c) Iba1
immunofluorescence (green) ipsilateral to DPI, (d) Iba1 immunofluorescence
contralateral to DPI. (e) Quantification of intensity of GFAP labeling intensity
from stained sections of trigeminal nucleus, averaged by animal and by side. Greater
fluorescent intensity was measured on the side ipsilateral to injury (two-way RM
ANOVA: Side: p = <0.0001, F = 62.7,
DOF = 1; Interaction: p = 0.2 (ns); Nucleus:
p = 0.5 (ns)). Bonferroni multiple comparison tests identified a
difference between the ipsilateral and contralateral sides in both the n. caudalis
(p < 0.001) and transition zone
(p < 0.01). (b) Quantification of intensity of Iba1 labeling
intensity, averaged by animal and by group assignment. Greater fluorescent intensity
was measured on the side ipsilateral to injury (two-way RM ANOVA: Side:
p = 0.01, F = 9.5, DOF = 1;
Interaction: p = 0.3 (ns); Nucleus: p = 0.9 (ns)).
Bonferroni multiple comparison tests only found a difference in sections from the
transition zone (p < 0.05).
Discussion
In this manuscript, we present the results of a series of experiments designed to identify neurobiological mechanisms that are initiated after DPI, and which could be involved in the establishment of chronic pain. Injury to the dental pulp is a common occurrence, either via bacterially mediated inflammation and eventual necrosis, or during dental procedures where the pulp is fully or partially removed. We found that some mechanisms identified in chronic neuropathic pain animal models also occur after DPI. These findings are important because they provide a biological basis for pain chronification after pulp injury.
Persistent orofacial pain that is not attributable to any apparent pathology in or around the tooth, and occurs after pulpal injury or surgery, is usually characterized as neuropathic pain.42,43 The fundamental requirement for pain to be characterized as neuropathic is identification of injury or lesion in the nervous system. 44 During pulpitis, the dental pulp tissue becomes inflamed, and if the source of inflammation is not removed, the highly innervated dental pulp degenerates. We assessed expression levels of the transcription factor ATF3 to detect peripheral nerve injury. We chose this marker for several reasons. In animal studies of disease and injury, including nerve transection/ligation, HIV, diabetic, and neurotoxin-induced neuropathies, ATF3 is used as a specific marker of nerve injury.17,45–47 Increased expression of ATF3 after peripheral nerve injury results from a loss of target tissue-derived growth factors. The upregulation in ATF3 then increases the growth state of dorsal root ganglion (DRG) neurons as part of the regenerative process for damaged afferents.48,49 Although increased expression of ATF3 is clearly a marker of peripheral nerve damage, its contribution to the establishment of post-injury hyperalgesia is unclear. That being said, ATF3 upregulation in injured DRG neurons is associated with the development of distinct pain states and was recently demonstrated to identify sensitized neurons after incision injury50,52 (but also see literature 51 ). Collectively, it appears that ATF3 is a sensitive marker of injured peripheral afferents and may well mark those neurons demonstrating prolonged sensitization after tissue injury. 52
In our model of a chronic progressive pulp injury, we observed a small but significant increase in ATF3 at the transcript and protein level in the TG ipsilateral to the injury. Although the magnitude of the increase was small, this is not surprising given that the injury only involved the pulp of two maxillary molar teeth. Changes in expression were assessed at the level of the whole TG, which contain the somatosensory cell bodies for the entire head and neck. Thus, the assessment of change in expression of ATF3 was inherently challenged by the small number of potentially injured neurons (those innervating only two teeth) relative to the abundance of uninjured neurons in the TG. Despite this we still observed a significant increase in ATF3 expression in the TG out to 28 days, suggesting that injured pulpal afferents engage signaling mechanisms similar to those occurring after injury to larger nerve branches.
The demonstration of increased ATF3 after pulp injury suggests that neuropathic mechanisms could be involved when persistent pain after pulp injury occurs. However, preclinical models of acute inflammatory pain have also demonstrated upregulation of ATF3 in DRG or TG as well as other classically “neuropathic type” changes in the peripheral and CNS.47,53 Because the distinction between inflammatory and neuropathic mechanisms have blurred as more preclinical pain models are characterized, we have chosen to use the language of “pathological” changes, as have others. The demonstration that ATF3 expression increases after DPI means that the pulp is not somehow protected from nerve injury signaling as occurs when larger nerve branches are injured (e.g., lingual, inferior alveolar nerve). It is feasible that pulpal afferents could be somehow protected from this type of signaling, given that they undergo painless programmed denervation when the primary set of teeth are exfoliated.54,55 Because nerve injury is a strong risk factor for developing chronic pain, and ATF3 expression in individual DRG neurons is associated with cellular hypersensitivity, the observed increase in ATF3 expression points to one possible mechanism by which DPI could lead to persistent pathological pain.52,56
We also observed an increase in ATF3 protein expression in a model of chronic neuropathic pain involving bilateral ligation of the mental nerve. However, we did not detect a significant increase in ATF3 mRNA. There are several possible explanations for this. We believe the most likely explanation is the limited number of injured afferents relative to the entire TG. In quantitating ATF3 protein expression, in which case we did see a significant increase in ATF3 after MNI, we focused our counting to the mandibular region of the TG, thus increasing the relative proportion of injured to uninjured afferents, essentially increasing the signal-to-noise ratio. On the other hand, RNA was extracted from the entire TG, greatly decreasing the relative proportion of injured to uninjured afferents. Another possibility is that we missed peak ATF3 gene expression at the time points we assessed the nerve injury. We chose an early (5 day) and a late (42 day) time point to assess expression. However, as ATF3 expression is readily detectable within 24 h after nerve injury, and levels are sustained for several weeks, the five-day time point should have included peak ATF3 expression.17,51
Despite the identification of increased nerve injury markers and glial upregulation in the brainstem trigeminal nucleus after DPI, we did not observe persistent hindpaw allodynia. The development of hindpaw allodynia has previously been reported to correlate with facial allodynia in rodent models of migraine.37,38 Given the difficulty of measuring mechanical withdrawal thresholds in the face of a mouse, this is a useful surrogate behavioral measurement for facial allodynia. We observed a transient hindpaw allodynia in mice with DPI, which resolved after 12 days, while mice with MNI had hindpaw mechanical allodynia that persisted out to 36 days. This suggests that some central mechanisms are engaged early after DPI but then resolve. It is possible that the first two weeks following DPI might represent the peak period of central neuroplasticity, as this is when most of the afferents in the central pulp would degenerate. However in rats, where it is more feasible to measure mechanical thresholds in the orofacial region, persistent allodynia in the extraoral tissues adjacent to the injured tooth was measured at least out to 28 days. 57 The mechanisms of extracephalic allodynia after orofacial injury or inflammation are not fully understood and warrant further study. We do not interpret the measurement of hindpaw allodynia as an indication of ongoing orofacial pain, but do think it reflects central neuroplasticity subsequent to DPI.
The identification of relevant, feasible, and robust rodent behavioral measures of orofacial pain remains a major challenge in the field. The measurement of mechanical withdrawal threshold as a primary behavioral outcome has been criticized because it captures a reflexive withdrawal and does not account for spontaneous ongoing pain. Thus, the results of analgesic efficacy in preclinical pain models relying on such behavioral outcomes may be misleading.58,59 We have previously explored several behavioral assays to better capture spontaneous orofacial pain, including home cage monitoring experiments and voluntary sucrose consumption. 22 We previously observed that mice with chronic DPI increase their consumption of sucrose solution, for more than 20 days after injury. In this study, we again observed a long lasting increase in consumption of a sucrose solution after DPI. However, in mice with mental nerve ligation, a model of chronic orofacial neuropathic pain, there was no difference between nerve injured and sham mice in the sucrose consumption assay. This suggests that the utility of the sucrose consumption assay is injury dependent. Other studies evaluating consumption of palliative substances after injury confirm that the utility of the assay needs to be validated for different injury or disease models. For example, in an open, brightly lit testing chamber, in which rodents are generally averse to spending time in the center, when palliative treats are placed in the center of the test chamber, mice with a painful paw incision consume more treats and spend more time in the center of the test chamber than uninjured mice. 60 On the other hand, a study using a bottle preference paradigm, with one bottle containing a palliative saccharine solution, and the other water, mice experiencing acute abdominal pain consumed proportionally less saccharine water than control mice. 61 Taken together, it appears that rodent assays assessing the impact of pain on motivational/reward driven behaviors warrant further study, but need to be carefully validated for different pain models, and specific testing paradigms.
We identified another limitation to the utility of the sucrose consumption assay in this paper. We attempted to reverse the increase in sucrose consumption after DPI with gabapentin. The experiment was done 21 days after DPI, when DPI mice still demonstrated increased sucrose consumption relative to sham mice. We observed that DPI mice decreased their sucrose consumption whether they received gabapentin or vehicle treatment (Supplementary Figure 2). This was observed in two independent experiments in which mice were carefully acclimated for multiple days to receive the drugs via i.p. injection. Our interpretation of this finding was that despite the attempts at acclimation, the stress produced by the injection influenced the sucrose consumption, masking any potential analgesic efficacy of the drug. Interpretation of results from assays such as the sucrose consumption assay, which are attempting to measure the affective-motivational state of rodents, is complicated by the involvement of high level brain function and all its intricacies.62,63 The use of catheters or mini-osmotic pumps, which would allow the non-invasive administration of drugs, should improve the usefulness of such assays in preclinical pain studies. At this point, we have only been able to successfully test immediate post-operative analgesics in the sucrose consumption assay, as reported in a prior publication. 22
Finally, we observed a significant upregulation of the microglia and astrocyte markers Iba1 and GFAP in the brainstem, ipsilateral to DPI. Although to our knowledge this is the first demonstration of glial upregulation in the medullary dorsal horn after pulp injury, the finding is congruous with other published studies. Gobel and Binck 64 studied CNS effects of pulp injury and described the involvement of glia in the removal of axonal debris from degraded pulpal afferent terminals in the outer lamina of the trigeminal nucleus. Other studies have reported increased GFAP expression in the TG after pulp exposure.65,66 Finally, in a model of acute pulpal inflammation with mustard oil, glial inhibitors blocked central sensitization. 67 Signaling between glia and neurons in the CNS play an important role in the development of pathological pain processes, including central sensitization and neuropathic pain, and therapeutics that interfere with this signaling are being pursued as novel analgesics to treat chronic pain.68,69 The demonstration of increased glial markers in this study suggests that potentially pathologic CNS plasticity occurs in response to pulp injury, providing a mechanism for persistent post-treatment pain in dentistry. Further studies exploring the role of glia-neuron interaction in the TG and medullary dorsal horn would be well worth pursuing.
A limitation of our model is that pulp injury was produced by creating a defect in the tooth that allowed oral pathogens access to the dental pulp, causing tissue necrosis, without an intervening treatment to resolve the inflammation. In patients, the pulp infection would usually be resolved by root canal treatment or tooth extraction. A small percentage of those patients, after seemingly successful treatment, will experience persistent pain. Although those patients did indeed experience DPI at some point in time, the factors leading to pain chronicity are multiple and complex and not fully captured in the DPI mouse model. Comparable limitations to interpreting findings in the DPI model exist in interpreting findings from rodent models of nerve ligation injury, such as CCI, and relating this to chronic post surgical pain. These limitations should be considered when interpreting these findings.
In conclusion, these studies describe several potential mechanisms whereby neuroplasticity in the peripheral and CNS after DPI could contribute to the development of chronic pain. The persistence of pain after seemingly successful dental interventions is perplexing to clinicians and causes pain and psychological suffering in patients. Further studies are needed to improve our understanding of these processes and to identify interventional strategies to either prevent the transition form acute to chronic pain or alleviate symptoms of established chronic pain.
Footnotes
Author Contributions
JLG: designed the study; JLG and HBG: performed experiments; AAR, CSL, HBG, EP, and JM: collected data; JLG, AAR, CSL, and JM: analyzed data; JLG and BM: wrote the manuscript. CSL and AAR: contributed equally to the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from NIH/NIDCR [Grant # K23DE019461, R03DE023153].
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.