
Abstract
Background
Gαi-interacting protein (GINIP) is expressed specifically in dorsal root ganglion (DRG) neurons and functions in modulation of peripheral gamma-aminobutyric acid B receptor (GBR). Genetic deletion of GINIP leads to impaired responsiveness to GBR agonist-mediated analgesia in rodent. It is, however, not defined whether nerve injury changes GINIP expression.
Results
Immunolabeling with validated antibody revealed GINIP expression in ∼40% of total lumbar DRG neurons in normal adult rats. GINIP immunoreactivity was detected in ∼80% of IB4-positive (nonpeptidergic) and ∼30% of CGRP-positive (peptidergic) neurons. GINIP immunoreactivity in the spinal cord dorsal horn was colabeled with IB4 and partially with CGRP. In addition, GINIP was expressed in DRG neurons immunopositive for GBR1, GBR2, Gαi(s), and Gαo and was also extensively colabeled with multiple nociceptive neuronal markers, including Trpv1, NaV1.7, CaV2.2α1b, CaV3.2α1b, TrkA, and Trek2. Peripheral nerve injury by L5 spinal nerve ligation significantly decreased the proportion of GINIP immunoreactivity-positive neurons from 40 ± 8.4% to 0.8 ± 0.1% (p < 0.01, mean ± SD, four weeks after spinal nerve ligation) and the total GINIP protein to 1.3% ± 0.04% of its basal level (p < 0.01, n = 6 animals in each group, two weeks after spinal nerve ligation) in the ipsilateral L5 DRGs.
Conclusion
Our results show that GINIP is predominantly expressed by small nonpeptidergic nociceptive neurons and that nerve injury triggers loss of GINIP expression. Signal transduction roles of GINIP may be diverse as it colabeled with various subgroups of nociceptive neurons. Future studies may investigate details of the signaling mechanism engaged by GINIP, as well as the pathophysiological significance of lost expression of GINIP in neuropathic pain.
Keywords
Background
Sensory impulses triggered by noxious stimuli are delivered to the dorsal horn (DH) of spinal cord via primary sensory neurons whose somata are located in the dorsal root ganglia (DRG) and cranial ganglia. Damage to peripheral nerves often induces a variety of molecular changes that lead to hyperexcitability and ectopic firing of primary sensory neurons, as well as sensitization of DH sensory neurons, which combined contribute to neuropathic pain.1–4
Metabotropic gamma-aminobutyric acid (GABA) type-B receptors (GBRs) are heterodimeric G-protein-coupled receptors (GPCRs) for GABA, the main inhibitory neurotransmitter.5,6 Appropriate GABAergic inhibition in the pain pathways plays fundamental roles for normal nociception. GBRs that have long been known to mediate analgesia in the central nervous system7,8 are also robustly expressed in peripheral sensory neurons positioned on the neuronal somata and presynaptic inhibitory terminals,9–15 and GABAB currents are identified in the DRG neurons. 16 In addition, a marked decrease in GABA sensitivity has been reported in dorsal roots after axotomy 17 and mice lacking functional GBRs exhibit pronounced hyperalgesia,18,19 corroborating a role for GBRs in nociception. However, the mechanism of GBR regulation in the peripheral sensory pathway and role of peripheral GBRs in the genesis of neuropathic pain is largely unknown.
Heterotrimeric G proteins are well known to act as intracellular transducers to propagate a variety of signals across the plasma membrane, 20 and GBRs activate Gαi/o-type G-proteins that inhibit adenylyl cyclase via Gαi/o and modulate ion channels via Gβγ.6,21 A recent study identified a novel Gαi-interacting protein (GINIP), also known as Kiaa1045 and encoded by the Kiaa1045 gene, which is expressed selectively in nociceptive sensory neurons.22,23 A physical interaction between GINIP and Gαi was defined, demonstrating GINIP is coupled to Gαi signaling pathway. Mice null for GINIP develop a selective and prolonged mechanical hypersensitivity after peripheral inflammation and neuropathy, with impaired responsiveness to baclofen, a GBR agonist, but not to delta or mu opioid receptor agonist-mediated analgesia in the spared nerve injury (SNI) model of neuropathic pain. GINIP-null DRG neurons exhibit deficient baclofen-evoked inhibition of high-voltage-activated calcium channels, and such mice show defective presynaptic inhibition of lamina II interneurons in the DH. 22
GINIP acts as an important nociceptor-specific modulator of GBRs in the peripheral sensory pathways. 22 It is, however, not defined whether peripheral nerve injury induces changes in GINIP expression. In this study, we characterized GINIP protein expression in the setting of nerve injury-induced pain. Our findings suggest that GINIP is particularly expressed in small nonpeptidergic nociceptive neurons and also that nerve injury triggers loss of GINIP expression.
Methods
Animals
Male Sprague Dawley rats (5–6 weeks old; 125–150 g body weight) were purchased from Charles River Laboratories (Wilmington, MA). All animal procedures were reviewed and approved by the Animal Care Committee of the Zablocki VA Medical Center Animal Studies Subcommittee and Medical College of Wisconsin IACUC (Permission number: 3690-03). Rats were housed in standard 12-h cycle lighting and were allowed ad libitum access to food and water prior to and throughout the experimental protocol.
Immunohistochemistry and quantification
Primary antibodies and IB4 used for IHC in this study.

GINIP: Gαi-interacting protein.
aAntibody abbreviations: Tubb3: β3-Tubulin; NF200: Neurofilament 200; CGRP: Calcitonin gene related peptide; Gαi1: G protein G(i) subunit alpha-1; Gαo: G protein G(o) subunit; GBR1: GABAB receptor 1; GBR2: GABAB receptor 2; Trpv1: Vanilloid receptor 1; NaV1.7: Voltage-gated sodium channel 1.7; CaV2.2: VGCC, N type, alpha 1B subunit; CaV3.2: VGCC, T type, alpha 1B subunit; TrkA: Tyrosine kinase receptor type A; Trek2: K channel, subfamily K, member 10.
bIt is an affinity purified goat polyclonal antibody raised against a peptide mapping near the N-terminus of GINIP of human origin.
cSCB, Santa Cruz Biotechnology, Santa Cruz, CA; Sigma–Aldrich, St. Louis, MO; ThermoFisher, Waltham, MA; Neuromics, Edina, MN; Alomone Labs, Jerusalem, Israel; Atlas Antibodies, Stockholm, Sweden.
For quantification of profiles of GINIP-positive DRG neurons, sections were immunostained for GINIP and counterstained with β3-tubulin (n = 6 DRGs, L4/L5), as well as different markers as indicated. IHC pictures were selected from different levels (usually a 10-section interval). The intensity of GINIP immunoreactivity (IR) in neurons higher than mean plus two folds of SD of the soma of negative neurons from each section was considered positive. Percentages of GINIP-positive neurons were obtained by counting the number of GINIP-positive cells and total positive cells of a given marker. All of the counting, including the quantification of colocalization, was performed using Adobe Photoshop CS6 software. The cross-sectional area and intensity (mean gray value) of GINIP-positive neurons were also collected using ImageJ v.1.46 software (National Institutes of Health). The background for each ganglion was subtracted for intensity quantification. Size distribution of GINIP-positive DRG neurons was performed according to the method as prior described25,26 using size cutoff classification criteria in rat as follows: small (<700 µm2), medium (700–1500 µm2), and large (>1500 µm2). For the quantification of colocalization between GINIP and markers, three to five slides for each marker (n = 3–6 DRGs) were double stained, images captured, and neuronal profiles counted using Photoshop CS6.
Experimental peripheral nerve injury and behavioral testing
Peripheral nerve injury was induced in isoflurane-anesthetized animals with tight ligation of the right L5 spinal nerve between DRG and the beginning of the spinal nerve, and behavioral tests were carried out as previously described. 27 Mechanical withdrawal threshold testing (von Frey) was performed using calibrated monofilaments (Patterson Medical, Bolingbrook, Illinois, USA). Briefly, beginning with the 2.8 g filament, filaments were applied with just enough force to bend the fiber and held for 1 s. If a response was observed, the next smaller filament was applied, and if no response was observed, the next larger was applied, until a reversal occurred, defined as a withdrawal after a previous lack of withdrawal, or vice versa. Following a reversal event, four more stimulations were performed following the same pattern. The forces of the filaments before and after the reversal, and the four filaments applied following the reversal, were used to calculate the 50% withdrawal threshold. Rats not responding to any filament were assigned a score of 25 g. Noxious punctate mechanical stimulation (pin test) was performed using the point of a 22 g spinal anesthesia needle that was applied to the center of the hindpaw with enough force to indent the skin but not puncture it. Five applications were separated by at least 10 s, which was repeated after 2 min, making a total of 10 touches. For each application, the induced behavior was either a very brisk, simple withdrawal with immediate return of the foot to the cage floor, or a sustained elevation with grooming that included licking and chewing, and possibly shaking, which lasted at least 1 s. This hyperalgesic behavior is specifically associated with place avoidance. 28 Hyperalgesia was quantified by tabulating hyperalgesia responses as a percentage of total touches.
Immunoblot analysis
DRG protein homogenates were extracted using 1 × RIPA buffer (20 mm Tris-HCl pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS, with 0.1% Triton X100 and protease inhibitor cocktail). As a positive control for GINIP expression, 293 T cells transfected with the plasmid pCMV-GINIP containing Flag and Myc tag at C-terminus (RC212001, Origene, Rockville, MD) was extracted at the same time. Protein concentration determined by using the BCA kit (Pierce, Rockford, IL). DRG homogenate and 293 T lysates protein (20 µg) were size separated using a 4%–12% gradient SDS-PAGE gel, transferred to 0.22 µm nitrocellulose membrane, and blocked in 5% skim milk. The blots were subsequently incubated overnight at 4℃ with a polyclonal goat anti-GINIP antibody (1:400; Santa Cruz Biotechnology (SCB)). Immunoreactive proteins were detected by enhanced chemiluminescence (Pierce, Rockford, IL) after incubation with HRP-conjugated anti-goat IgG (1:2000, SCB). The same membrane was stripped at 60℃ for 30 min with stripping buffer containing 62.5 mm Tris-HCl, pH 6.8, 2% SDS, 100 mm 2-mercaptoethanol and reprobed with anti β-actin (1:1000, SCB) used for loading control. Densitometry of bands of interests was analyzed using ImageJ v.1.46. Ratios of the band density of GINIP to β-actin were calculated and the percentage changes of GINIP in the experimental samples compared with those from the control samples.
Statistics
Statistical analysis was performed with GraphPad PRISM (GraphPad Software, San Diego, CA). Behavioral changes over baseline for von Frey measurements were made using repeated measures one-way ANOVA and post hoc analysis with Bonferroni test, and for pin test using non-parametric analysis with post hoc paired comparison by Dunn’s test. In vivo GINIP expression in DRGs was assessed by one-way ANOVA and post hoc analysis with Tukey’s test. Results are reported as mean and standard deviation (SD). p < 0.05 were considered statistically significant.
Results
GINIP is abundantly expressed in DRG nociceptive neurons
The first set of studies examined GINIP expression in lumbar DRGs of control adult rat by IHC. GINIP was found in ∼40% of Tubb3 (a pan DRG neuronal marker) stained neuronal profiles (Figure 1(a)). No staining was evident in sections preincubated with the corresponding antigen peptide (data not shown), validating the specificity for the staining patterns obtained with this antibody. To determine the phenotype of neurons that express GINIP, we used the common nonpeptidergic marker isolectin B4 (IB4), peptidergic marker calcitonin gene-related peptide (CGRP), and neurofilament 200 (NF200), a marker for myelinated Aδ and Aβ neurons. An average of 80% of GINIP-positive neurons binds IB4 and 82% of IB4-positive neurons expresses GINIP. On average, 30% of GINIP-positive neurons express CGRP while 30% of CGRP-positive neurons express GINIP. Some GINIP neurons are positive for both CGRP and IB4, and we found an overlap (∼30%) of CGRP-expressing neurons with IB4 binding (CGRP/IB4 double positive, data not shown). NF200-positive neurons are generally larger than CGRP- and IB4-positive neurons, and most do not display colocalization with GINIP (Figure 1(b)–(d)). Overall, GINIP appeared as variably intense cytoplasmic immunopositivity, mainly in small-sized neurons, with some low-intensity staining in medium-sized neurons, while no GINIP IR was detected in large-sized neurons. These results indicate that GINIP is predominantly expressed in the non-myelinated C- or lightly myelinated Aδ-fiber nociceptive neuron populations.
Expression of GINIP and colocalization with classic markers in lumbar DRG and SC. (a) Immunostained GINIP profile with Tubb3 counterstaining of all neurons in DRG section. (b) IHC images show colocalization of GINIP-positive neurons with CGRP, IB4, or NF200. (c) Percentage colocalization of GINIP with Tubb3, CGRP, IB4, and NF200. (d) Relative fluorescence intensity plotted vs. cross sectional area of GINIP-positive neurons. Dashed line indicates cutoff level of background signals. (e) GINIP immunopositivity in the superficial layers of spinal dorsal horn (white dashed line outlines gray matter in a cross-section of lumbar spinal cord with white matter blue pseudocolored). (f) GINIP signals colocalize mostly on IB4-positive laminae II afferents and (g) some GINIP signals colocalize with CGRP-positive central terminal fibers. Scale bars: 50 µm for all.
GINIP is transported to central presynaptic terminals
In the lumbar spinal cord, GINIP was symmetrical in control rats (Figure 1(e)), and mostly concentrated on IB4-positive laminae II afferents (Figure 1(f)). GINIP co-staining was evident in some CGRP-positive presynaptic terminal fibers (Figure 1(g)), but no GINIP-positive fibers were seen in the other spinal lamina and the cells of spinal cord did not express GINIP. These results indicate that GINIP is synthesized in the cell body of DRG neurons and transported to their axon central terminals in DH superficial laminae. When the sciatic nerve was stained in naive animals, only faintly stained GINIP-positive fibers were observed (data not shown). No GINIP-positive fibers were found in the skin sections of the hind paw. Taken together, GINIP is centrifugally transported but their levels in peripheral skin are mostly undetectable with our technique.
Colocalization of GINIP with multiple distinct subpopulations of DRG neurons
GINIP is a putative modulator of GBRs that function by coupling to Gαi/Gαo-mediated intracellular signaling cascades.
29
We next performed double immunostaining for the colocalization of GINIP with Gαi, Gαo, GBR1, and GBR2 in lumbar DRG sections from naïve rats. To define Gαi neurons, we used a Gαi1 antibody that predominantly recognizes Gαi1, and also, to a lesser extent, Gαi2 and Gαi3, according to the manufacturer. The presence of Gαi, Gαo, GBR1, and GBR2 IR was observed in the majority of DRG neurons, with higher IR intense in smaller diameter cells. About 40% of Gαi1- or Gαo-positive, and ∼60% of GBR1- or GBR2-positive neurons express GINIP, and virtually the majority of GINIP-positive cells express Gαi1, Gαo, GBR1, or GBR2, respectively (Figure 2(a)–(c)).
Colocalization of GINIP with Gαi, Gαo, and GBRs. (a) Double labeling of GINIP with Gαi, Gαo, GBR1, and GBR2, labeled on the top with staining in the left of panels. (b, c) Bar charts illustrate percentage colocalization of GINIP with each marker. Scale bar: 50 µm for all.
Inhibitory GPCRs, including GBRs, are distributed among nociceptors.11–13,15,30,31 We, therefore, further examined the expression of GINIP relative to various molecules that are known to be expressed in different subpopulations of nociceptive neurons, using a panel of markers including Trpv1, NaV1.7, CaV2.2α1b, CaV3.2α1b, TrkA, and Trek2 (Figure 3(a)–(c)). Overall, about half of neurons positive for each marker coexpress GINIP, and nearly all of the GINIP-positive neurons coexpress those markers. Collectively, these histological findings reinforce that GINIP is expressed in sensory neurons with nociceptive features.
Colocalization of GINIP with subpopulations of DRG nociceptive neurons. (a) Double labeling of GINIP with multiple authentic nociceptive neuronal markers (Trpv1, NaV1.7, CaV2.2α1b, CaV3.2α1b, TrkA, and Trek2), labeled on the top with staining in the left of panels. (b, c) Bar charts illustrate percentage colocalization of GINIP with each marker. Scale bar: 50 µm for all.
Axotomy diminishes GINIP protein expression
Having established the GINIP profile in naïve rats, we next sought to examine alteration of GINIP expression by peripheral injury. For this, we used L5 spinal nerve ligation (L5 SNL), a well-established animal model of chronic neuropathic pain. Following SNL, behavioral testing revealed mechanical hypersensitivity. Specifically, reduced threshold for withdrawal from mild mechanical stimuli (von Frey testing, Figure 4(a)) represents the development of mechanical allodynia, and exaggerated and sustained responses to fully noxious mechanical stimulation (Pin testing, Figure 4(c)) represents the development hyperalgesia. These changes lasted throughout the four-week testing interval.
GINIP expression in SNL rats. Rats (n = 5) with L5 SNL injury developed significant mechanical allodynia (von Frey, a) and hyperalgesia (Pin, c). (b, d) The GINIP antibody recognized an expected band (∼50 kDa) in the Western blots (Wb) from 293 T cells heterogeneously expressing GINIP (lane 12) or from the rat DRG homogenates (lanes 1–9) but did not recognize GINIP in the control 293 T cell lysate (lane 11) or empty lane (lane 10). At three days after L5 SNL, GINIP protein decreased to 5.3 ± 4.3% of the level in the ipsilateral L5 (iL5) DRGs and 69.2 ± 6% in non-injured ipsilateral L4 (iL4) DRGs of the basal level in the contralateral L5 (cL5) DRGs (***p < 0.001, n = 3 animals in each group) (b). At two weeks after L5 SNL, GINIP protein was barely detectable in the iL5 DRGs and total GINIP protein dropped to 1.3 ± 0.1% of its basal level in the cL5 DRGs (***p < 0.001, n = 6 animals in each group). No significant change of GINIP protein level in non-injured iL4 DRGs was observed compared to cL5 DRGs (d). (e) IHC shows normal profiles of GINIP expression in naïve L5 and non-injured iL4 DRGs while GINIP is barely detectable in the axotomized iL5 DRGs with diminished IB4 binding (four weeks after L5 SNL). (f) Four weeks after SNL, GINIP IR is mostly eliminated in ipsilateral DH superficial layers parallel with decreased ipsilateral IB4 and CGRP staining. Scale bars: 50 µm for all.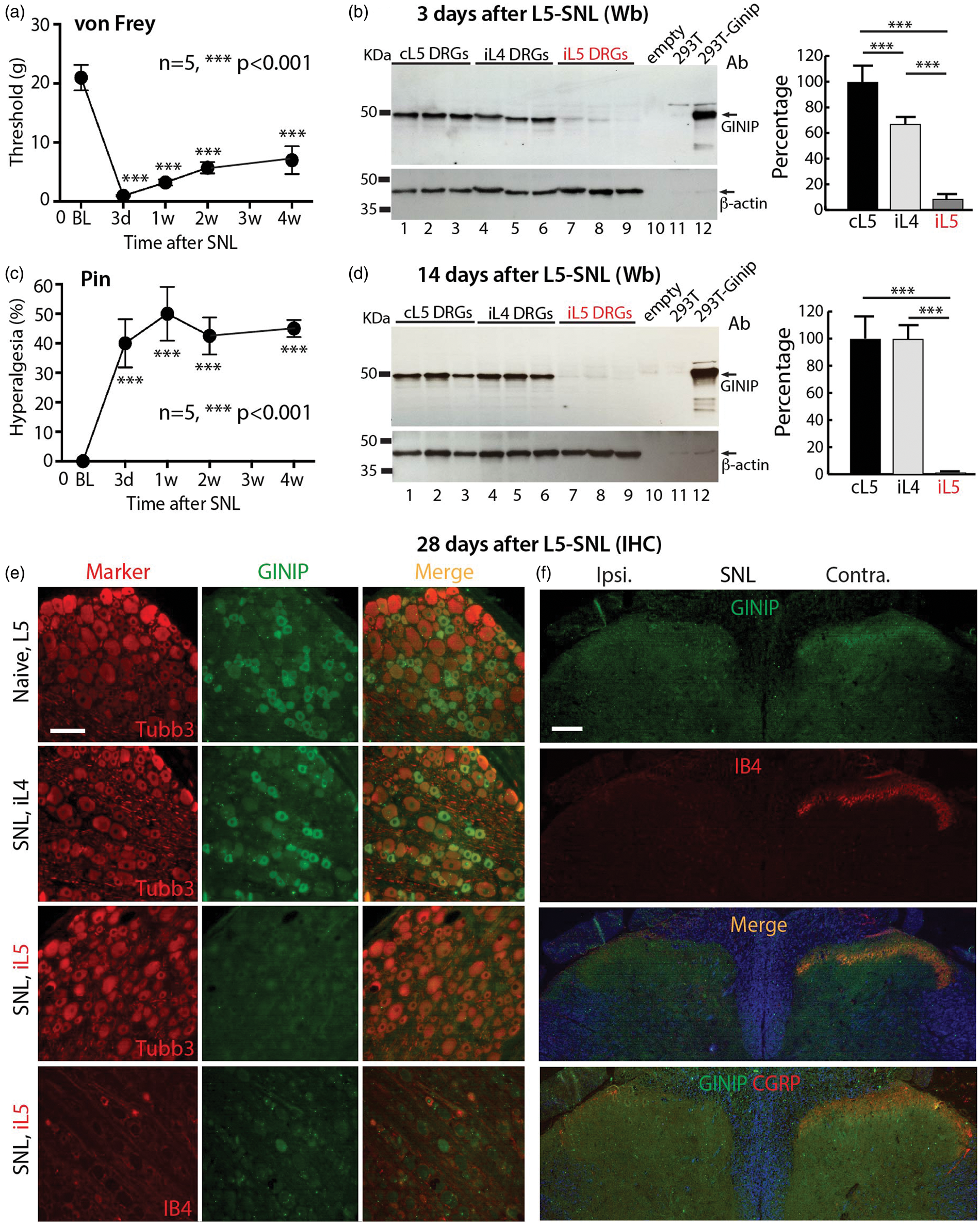
Western blots of naïve DRG homogenates revealed a strong band at ∼50 kDa, which aligns with the size of the positive band in the lysates from GINIP-cDNA transfected 293 T cells (Figure 4(b), (d)). In the early stage following axotomy, three days after injury, GINIP protein in the ipsilateral L5 DRGs decreased to 5.3 ± 4.3% of the level in the contralateral L5 DRGs, while GINIP level in neighboring uninjured L4 DRGs dropped to 69.2 ± 6% of the contralateral level (n = 3 animals in each group, mean ± SD; Figure 4(b)). Two weeks after L5 SNL, GINIP protein was barely detectable in the ipsilateral L5 DRGs, at a level 1.3 ± 0.1% of that in the contralateral L5 DRGs (n = 6 animals in each group; Figure 4(d)). No significant change was observed in the GINIP protein level in non-injured ipsilateral L4 DRGs at two weeks after SNL, compared to the contralateral DRGs.
IHC analyses were performed on DRG and SC sections harvested at four weeks following SNL. This showed that the proportion of GINIP-positive neurons significantly decreased from 40 ± 8.4% to 0.8 ± 0.1% (p < 0.01, n = 4 animals in each group, mean ± SD) in ipsilateral L5 DRGs (Figure 4(e)). In parallel, GINIP IR dramatically diminished in ipsilateral DH in SNL rats (Figure 4(f)). Injury-induced loss of GINIP was concurrent with decreased CGRP staining and loss of IB4 binding, which became barely detectable in ipsilateral DH at spinal cord levels corresponding to the axotomized DRG. No changes were observed in the adjacent non-injured L4 DRGs.
Together, peripheral nerve axotomy resulted in GINIP loss quickly after pain onset, and near completely loss of GINIP was evident at two and four weeks after injury. GINIP was also moderately and temporarily reduced in the acute stage in the adjacent non-injured L4 DRGs. At the behavioral level, GINIP loss was correlated temporally with development of pain behavior, including mechanical allodynia and hyperalgesia that were evident three days after injury and persisted during the four weeks of testing.
Discussion
We have found that GINIP represents a nociceptive neuronal maker with predominant nonpeptidergic nature. Our results, however, show that GINIP is also expressed in a substantial group (∼30%) of peptidergic neurons. Importantly, the present study demonstrates that GINIP protein is nearly depleted in DRG neurons whose axons are injured by peripheral nerve transection. Loss of GINIP in DRG neurons occurs rapidly after peripheral axotomy that leads to neuropathic pain and is temporally matched with the development of pain behavior in nerve-injured rats.
A previous study has reported that GINIP exhibits positive regulation for the Gαi-coupled GBRs in primary sensory neurons. 22 Thus, it is possible that injury-induced GINIP depletion may reduce GABAergic function in DRG neurons, leading to an attenuation of signaling via Gαi-coupled GBR pathways that mediate pain-inhibitory mechanisms. 32 In addition, GINIP is preferably expressed in non-peptidergic nociceptive neurons. These afferents terminate in the spinal superficial laminae, primarily in complex synaptic glomeruli and are postsynaptic to inhibitory GABAergic and glycinergic interneurons.33–35 This suggests that GINIP in the central terminals may serve to modulate inhibitory integration of the first sensory synapse between primary afferent and DH neurons, 22 and therefore, disruption or reduction of spinal cord nociceptive inhibitory gating due to loss of presynaptic GINIP could be expected.
Peripheral axotomy alters expression of many genes in primary sensory neurons, which play essential roles in contributing to neuropathic pain. 36 However, the factors that trigger GINIP depletion after nerve injury in the primary sensory neurons are unclear. One of the possibilities may be death of the axotomized neurons as IB4-binding is also lost. Indeed, peripheral nerve injury in neonatal rats results in the death of the majority of the axotomized sensory neurons by seven days after injury. However, in adult animals, most sensory neurons survive for at least four months after peripheral axotomy. 37 Additionally, IB4 binding lost after peripheral injury can be subsequently restored in both the DRG neurons and their central terminals in the spinal cord.25,38 These results suggest that the loss of GINIP immunoreactivity after axotomized injury does not represent a loss of neurons but instead simply a turning-off the protein expression by an unknown mechanism. As a limitation of the current study, we investigated the changes in GINIP protein expression only and did not determine GINIP transcription. Further study is needed to determine if GINIP is regulated after nerve injury at the transcriptional or translational level.
Notably, we found that GINIP coexpresses with multiple nociceptive proteins. This could be an immunohistochemical coincidence but may serve as an indication of a suspected physical and functional interaction since GINIP is potentially involved in many neural interactomes. 22 Therefore, whether there exist functional link between GINIP and these various nociceptive molecules warrants further investigation.
In summary, we show that peripheral nerve injury is associated with depletion of DRG nociceptor-specific GINIP, and GINIP loss is correlated temporally with development of pain behavior. Future studies may investigate details of the signaling mechanism engaged by GINIP, including the effects of GINIP on signal-receiving GABA receptors, transducing heterotrimeric G protein (Gαβγ subunits), and potential downstream target effectors. These investigations will provide insights into the pathophysiological significance of lost expression of GINIP in neuropathic pain.
Footnotes
Authors’ contributions
HY and QHH conceived and designed the experiments; ZL, HY, FW, and GF performed the experiments; ZL and HY analyzed the data; HY and QHH wrote the paper.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded in part by the VA Rehabilitation Research and Development Grant (I01RX001940, QHH) and Advancing a Healthier Wisconsin Grant (5520293, QHH and HY). ZL was a recipient of Chinese Scholarship Council.