
Abstract
Background
Glia–neuron interactions play an important role in the development of neuropathic pain. Expression of the pro-inflammatory cytokne →cytokine Interferon-gamma (IFNγ) is upregulated in the dorsal horn after peripheral nerve injury, and intrathecal IFNγ administration induces mechanical allodynia in rats. A growing body of evidence suggests that IFNγ might be involved in the mechanisms of neuropathic pain, but its effects on the spinal dorsal horn are unclear. We performed blind whole-cell patch-clamp recording to investigate the effect of IFNγ on postsynaptic glutamate-induced currents in the substantia gelatinosa neurons of spinal cord slices from adult male rats.
Results
IFNγ perfusion significantly enhanced the amplitude of NMDA-induced inward currents in substantia gelatinosa neurons, but did not affect AMPA-induced currents. The facilitation of NMDA-induced current by IFNγ was inhibited by bath application of an IFNγ receptor-selective antagonist. Adding the Janus activated kinase inhibitor tofacitinib to the pipette solution did not affect the IFNγ-induced facilitation of NMDA-induced currents. However, the facilitatory effect of IFNγ on NMDA-induced currents was inhibited by perfusion of the microglial inhibitor minocycline. These results suggest that IFNγ binds the microglial IFNγ receptor and enhances NMDA receptor activity in substantia gelatinosa neurons. Next, to identify the effector of signal transmission from microglia to dorsal horn neurons, we added an inhibitor of G proteins, GDP-β-S, to the pipette solution. In a GDP-β-S–containing pipette solution, IFNγ-induced potentiation of the NMDA current was significantly suppressed after 30 min. In addition, IFNγ-induced potentiation of NMDA currents was blocked by application of a selective antagonist of CCR2, and its ligand CCL2 increased NMDA-induced currents.
Conclusion
Our findings suggest that IFNγ enhance the amplitude of NMDA-induced inward currents in substantia gelatinosa neurons via microglial IFNγ receptors and CCL2/CCR2 signaling. This mechanism might be partially responsible for the development of persistent neuropathic pain.
Keywords
Background
Patients with neuropathic pain experience spontaneous tingling sensations, hyperalgesia, and tactile allodynia, symptoms that deteriorate their quality of life. The pathogenesis of neuropathic pain is believed to involve central sensitization of spinal dorsal horn neurons following central or peripheral nervous system damage.1–3 It was recently suggested that neuron–glia interactions play a crucial role in the development of central sensitization.4,5 Various cytokines or chemokines have been reported to be involved in facilitating neuron–glia crosstalk.6–8
Interferon-gamma (IFNγ) is a pro-inflammatory cytokine and a major modulator of central and peripheral immune responses 9 that plays roles in a variety of chronic pain conditions. For example, patients with persistent pain due to viral infections or multiple sclerosis have persistently high IFNγ levels in the spinal cord as an inflammatory response.10,11 This cytokine is produced primarily by T-cells and natural killer cells 12 and stimulates the Janus activated kinase (JAK)-signal transducer and activator of transcription (STAT) signaling pathway.13,14 Upon peripheral nerve injury, IFNγ is upregulated in the rat dorsal horn as T-cells that infiltrate the spinal cord, and it has been implicated as one of the causative agents of neuropathic pain.15–17 Interestingly, intrathecal injections of IFNγ induce pain-related behaviors in wildtype rats and mice but not IFNγ receptor knock-out mice.18–21 However, little is known about the roles of IFNγ in the dorsal horn of the spinal cord.
The substantia gelatinosa (SG, lamina II) of the spinal cord dorsal horn has been implicated in nociceptive transmission because both finely myelinated (Aδ) and unmyelinated (C) primary nociceptive afferent fibers are heavily projected to this spinal region.22–24 To elucidate how IFNγ affects nociceptive responses in the spinal dorsal horn, we analyzed the effects of IFNγ on postsynaptic action by using whole-cell patch-clamp recordings from SG neurons.
Methods
Five to six-week-old male Sprague Dawley rats (180–210 g; obtained from SLC, Japan) were used. The protocol was approved by the Ethics Committee on Animal Experiments, Wakayama Medical University, and was in accordance with the UK Animals (Scientific Procedures) Act of 1986 and associated guidelines. Animals were housed in plastic cages at room temperature on a 12-h light/dark cycle (light on between 8:00 a.m. and 8:00 p.m.) with ad libitum access to food and water.
Spinal cord slice preparation
L4-5 level transverse spinal cord slices were prepared, as described previously. 25 Briefly, the adult rats were anesthetized with urethane (1.2 g/kg, intraperitoneal), and Th10-L4 laminectomy was performed. Then, we excised the spinal cord and submerged it in preoxygenated Krebs’ solution at 1–3℃. Immediately after the removal of the spinal cord, the rats were killed by exsanguination under urethane. The dura, arachnoid, and pia mater were removed, and the spinal segment was sliced to 650-µm thickness by using a microslicer (DTK-1000, Dousaka from Kyoto, Japan). The slices were continuously perfused with Krebs’ solution (10 ml/min) and saturated with 95% O2 and 5% CO2. The perfusion solution was heated to 36 ± 1℃ by using a temperature controller. The Krebs’ solution contained the following (in mM): 117 NaCl, 3.6 KCl, 1.2 NaH2PO4, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, and 11 glucose, at pH 7.4.
Patch-clamp recordings from spinal cord slices
Blind whole-cell patch-clamp recordings were made from lamina II (SG) neurons in voltage-clamp mode. The patch-pipette had a resistance of 5–10 Ω, and the composition of the patch-pipette internal solution was as follows (in mM): 135 potassium gluconate, 5 KCl, 0.5 CaCl2, 2 MgCl2, 5 EGTA, 5 HEPES, and 5 ATP–Mg. Membrane currents were amplified by using an amplifier (Axopatch 200B from Molecular Devices, Sunnyvale, CA, USA) in voltage-clamp mode. Holding potential was set to −70 mV for recording exogenous α-amino-3-hydroxy-5-methyl-4-Isoxazole-4-propionic acid (AMPA) current and to −50 mV for N-methyl-
Drug application
The drugs used in this study were IFNγ (PeproTech, Rocky Hill, NJ, USA); IFNγ antagonist (VWR International, Radnor, PA, USA); monocyte chemoattractant protein 1 (MCP-1) (R&D, Minneapolis, MN, USA); and, AMPA, NMDA, minocycline hydrochloride, Teijin compound 1 hydrochloride, GDP-β-S, and tofacitinib citrate (Sigma–Aldrich, St. Louis, MO, USA). IFNγ antagonist, MCP-1, AMPA, NMDA, and minocycline hydrochloride were dissolved in distilled water as 1,000 × stock solutions, and Teijin compound 1 hydrochloride and tofacitinib citrate 26 were first dissolved in 5% DMSO as 1,000 × stock solutions. The 1,000 × stock solutions of IFNγ were prepared in distilled water containing 1% bovine serum albumin (BSA) (Sigma–Aldrich). All perfusion drugs were stored at −20℃ and diluted to the final concentration in Krebs solution just before use, and then were perfused via a three-way stopcock without any change in the rate or temperature. The time necessary for the solution to flow from the stopcock to the spinal cord section surface was about 1 min. GDP-β-S and tofacitinib citrate were added to the pipette solution. The tubing of our perfusion system was coated with AquaSil siliconizing fluid (Thermo Fisher Scientific, Waltham, MA, USA) to reduce the loss of IFNγ through nonspecific interactions.
Statistical analysis
All numerical data are expressed as mean ± standard error (SE). Paired Student’s t tests were used for statistical comparison, and the criterion for statistical significance was p < 0.05; n refers to the number of neurons studied.
Results
IFNγ enhanced NMDA-induced but not AMPA-induced inward currents
Only SG neurons, with resting membrane potentials lower than −50 mV in the current-clamp mode, were used for recording. Perfusion of exogenous AMPA (10 μM) for 30 s onto the spinal cord slice induces slow inward currents at a holding potential of −70 mV, and NMDA (50 μM) does so at −50 mV. This discrepancy is because NMDA receptors are blocked by Mg2+ when the cell membrane is held at −70 mV. AMPA superfusion repeatedly produces an inward current with the same peak amplitude in an SG neuron (101.3 ± 0.6%, n = 5, p = 0.09), observed for NMDA (97.9 ± 3.1%, n = 5, p = 0.53). We first examined the effects of IFNγ on inward currents induced by AMPA and NMDA when maintaining the voltage at −70 and −50 mV, respectively. Bath application of IFNγ (30 nM, 2.5 min) did not affect the AMPA-induced current (100.0 ±3.7%, n = 11, p = 0.87, mean amplitude of control: 51.5 pA; Figure 1(a),(c)). On the other hand, NMDA-induced current was significantly increased by IFNγ (147.3 ± 10.7%, n = 14, p = 0.0002, mean amplitude of control: 16.8 pA; Figure 1(b),(d)). This IFNγ-induced facilitation of NMDA current was inhibited by an IFNγ receptor-selective antagonist (300 nM, 5.5 min) (90.0 ± 3.6%, n = 10, p = 0.053; Figure 2). These results suggest that IFNγ binds to own receptor and enhances the activity of NMDA but not AMPA receptors in the dorsal horn of the spinal cord.
AMPA- and NMDA-induced currents under bath application of IFNγ in adult rat SG neurons. (a) Recordings of AMPA-induced inward currents at a holding potential of −70 mV. Perfusion of IFNγ (30 nM, 2.5 min) did not result in any significant change in AMPA-induced currents. (b) Effects of IFNγ on NMDA-induced inward currents at a holding potential of −50 mV. NMDA-induced current was significantly increased by IFNγ (30 nM, 2.5 min). (c) The peak amplitude of the AMPA-induced currents after IFNγ treatment was 100.0 ± 3.7% of baseline. (d) The peak amplitude of the NMDA-induced currents after IFNγ treatment was 147.3 ± 10.7% of baseline. In Figure 1(b) and subsequent figures, NMDA responses were recorded in voltage-clamp mode (VH = −50 mV). n.s.: not significant. * p < 0.05. AMPA: α-amino-3-hydroxy-5-methyl-4-Isoxazole-4-propionic acid; IFNγ: interferon-gamma; NMDA: N-methyl- An IFNγ receptor selective antagonist inhibited IFNγ-induced facilitation of the NMDA current. (a) Perfusion of the IFNγ receptor-selective antagonist (300 nM, 5.5 min) inhibited the IFNγ-induced facilitation of NMDA current. (b) Bar graphs show the peak amplitude of NMDA-induced current compared to baseline when the IFNγ receptor-selective antagonist and IFNγ are perfused simultaneously. Therefore, IFNγ likely binds to IFNγ receptors and activates NMDA receptors in SG neurons. IFNγ: interferon-gamma; NMDA: N-methyl-

Potentiation of NMDA-induced currents by IFNγ was mediated by glial activation
Because both neurons and microglia are reported as sites of IFNγ receptors,20,27 we investigated which receptor is responsible for the IFNγ-induced potentiation of NMDA-induced currents. As shown in Figure 3, the IFNγ-induced facilitation of NMDA currents was inhibited by minocycline (20 μM, 5.5 min), an inhibitor of microglia activation (90.3 ± 6.4%, n = 12, p = 0.12). In addition, we recorded the IFNγ-induced facilitation of NMDA current during inhibition of JAK-STAT signaling. We added the JAK inhibitor tofacitinib (1 mM) to the pipette solution. Despite blocking JAK-STAT signaling, IFNγ-induced facilitation of NMDA currents was not affected even after 30 min (98.9 ± 6.0%, n = 7, p = 0.99; Figure 4). Therefore, we considered that IFNγ activates the microglial IFNγ receptor and enhances the activity of NMDA receptors in the dorsal horn neuron.
The potentiation of NMDA-induced currents by IFNγ was inhibited by minocycline. (a) Minocycline (20 μM, 5.5 min), an inhibitor of microglia activation, inhibited the IFNγ-induced facilitation of the NMDA current. (b) Bar graphs show the peak amplitude of the NMDA-induced currents compared with baseline when minocycline and IFNγ were perfused simultaneously. Therefore, IFNγ likely binds to microglial IFNγ receptors and enhances the activity of NMDA receptors in dorsal horn neurons. IFNγ: interferon-gamma; NMDA: N-methyl- The JAK inhibitor tofacitinib had no effect on IFNγ-induced NMDA currents. (a) Tofacitinib (1 mM) addition to the pipette solution did not affect the IFNγ-induced NMDA current. In this experimental system, the control corresponds to the IFNγ-induced NMDA currents recorded immediately after patch clamping. (b) Bar graphs show the average rate of NMDA current enhancement by IFNγ at the start of patch-clamp recording and 30 min later. The peak amplitude of the IFNγ-induced NMDA currents did not change compared with the control recording. This confirmed that IFNγ enhances neuronal NMDA-induced inward current via the microglial IFNγ receptor. IFNγ: interferon-gamma; NMDA: N-methyl-

CCL2 is involved in IFNγ-induced activation of NMDA receptors
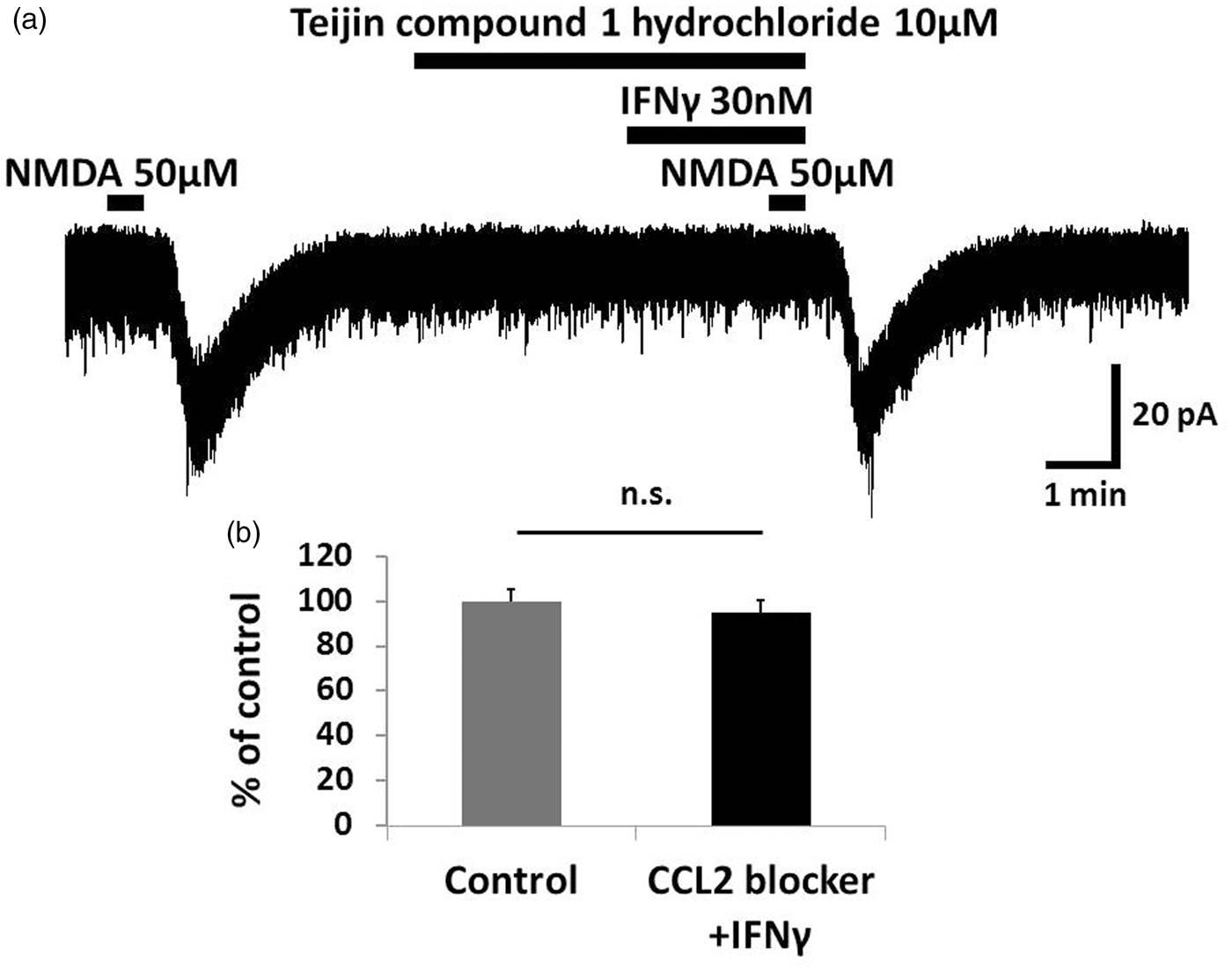
As JAK-STAT signaling was not the intraneuronal signal transducer underlying the effect of IFNγ on NMDA currents, we examined another possible signaling pathway. First, we added 1 mM GDP-β-S, a non-hydrolysable analog of GDP that competitively inhibits G proteins, to the pipette solution. NMDA-induced inward currents were significantly suppressed when IFNγ and NMDA were applied 30 min after patch clamping with pipettes containing GDP-β-S (78.7 ±5.1%, n = 6, p = 0.023; Figure 5). These findings suggested that the IFNγ-induced increase of NMDA currents involves the activation of G protein-coupled receptors such as chemokine receptors. Specifically, the chemokine MCP-1, also known as chemokine ligand 2, CCL2 and its receptor C-C chemokine receptor type 2 (CCR2) are reportedly involved in the generation of neuropathic pain4,5,7,28 and are related to IFNγ.9,29 We then investigated the participation of CCL2/CCR2 in this effect of IFNγ on NMDA currents. IFNγ-induced enhancement of NMDA currents was blocked by a selective antagonist of CCR2, Teijin compound 1 hydrochloride (10 μM, 5.5 min) (95.0 ± 5.3%, n = 6, p = 0.43; Figure 6). In addition, bath-applied CCL2 (100 ng/ml) for 2.5 min enhanced NMDA-induced inward current (128.3 ± 6.4%, n = 5, p = 0.006; Figure 7). Therefore, it is possible that microglia activated by IFNγ release CCL2, which binds to CCR2 and enhances neuronal NMDA-induced current.
The G protein inhibitor GDP-β-S significantly inhibited IFNγ-induced NMDA currents. (a) In a potassium gluconate pipette solution containing GDP-β-S (1 mM), the IFNγ-induced NMDA current after 30 min of recording was significantly lower than the control recording of IFNγ-induced NMDA currents recorded at the start of patch clamping. (b) Bar graphs show the average rate of IFNγ-induced enhancement of NMDA current in the same neuron at the beginning of the recording and after 30 min. The value of the right bar is 78.7 ± 5.1% of the left bar. This finding suggested that the IFNγ-induced enhancement of NMDA currents involves the activation of G protein-coupled receptors. *p < 0.05. IFNγ: interferon-gamma; NMDA: N-methyl- The enhancement of NMDA-induced inward current by IFNγ was mediated by CCR2. (a) Bath application of the CCR2 antagonist Teijin compound 1 hydrochloride (10 μM, 5.5 min) blocked the IFNγ-induced enhancement of NMDA currents. (b) The peak amplitude of the NMDA-induced currents after simultaneous application of Teijin compound 1 hydrochloride and IFNγ was 95.0 ± 5.3% that of baseline. IFNγ likely increases the NMDA-induced inward current in SG neurons via CCR2. IFNγ: interferon-gamma; NMDA: N-methyl- Bath application of CCL2 enhanced NMDA-induced inward current. (a) Perfusion of spinal cord slices with CCL2 (100 ng/ml, 2.5 min) increased NMDA-induced inward currents. (b) The peak amplitude of the NMDA-induced currents after simultaneous CCL2 application is 128.3 ± 6.4% compared with baseline. This suggests that IFNγ increases the NMDA-induced inward current in SG neurons via CCL2/CCR2 signaling. * p < 0.05. MCP-1: monocyte chemoattractant protein 1; NMDA: N-methyl-

Discussion
In the present study, IFNγ significantly enhanced NMDA-induced inward currents but not AMPA-induced inward currents in SG neurons. This IFNγ-induced facilitation of NMDA current was inhibited by an IFNγ receptor-selective antagonist. The results suggest that IFNγ enhances NMDA receptor signaling by activating IFNγ receptors in the dorsal horn of the spinal cord. Minocycline, an inhibitor of microglial activation, inhibited the IFNγ-induced facilitation of NMDA current, but inhibition of the neuronal JAK-STAT signaling did not. Therefore, we conclude that IFNγ binds its own receptors in microglia but not SG neurons, and the subsequent microglial activation leads to NMDA receptor activation. Moreover, IFNγ-induced enhancement of NMDA currents was blocked by GDP-β-S in the pipette solution or bath application of a CCR2 antagonist. CCR2 signaling is mediated by a G protein and a typical chemokine involved in central sensitization. Application of CCL2 alone also enhanced NMDA-induced current. These results suggested that activation of IFNγ receptors in the spinal microglia leads to CCL2 release and subsequently enhances NMDA receptors in dorsal horn neurons (Figure 8).
Schematic overview. When nerve injury occurs, IFNγ levels are increased in the dorsal horn of the spinal cord and binds to IFNγ receptors on microglia. CCL2 subsequently enhances the neuronal NMDA-induced inward current in lamina II neurons via CCR2. This potentiation of NMDA receptors likely contributes to the development of neuropathic pain. CCL2: chemokine ligand 2; CCR2: C-C chemokine receptor type 2; IFNγ: interferon-gamma; SG: substantia gelatinosa.
Importantly, this is the first study to investigate the function of IFNγ using blind whole-cell patch-clamp methods. Interestingly, inward currents induced by exogenously applied NMDA, but not AMPA, were clearly enhanced by the application of IFNγ in SG neurons of adult rat spinal cord slices. This postsynaptic effect of IFNγ on NMDA receptor could explain the pain-related behaviors caused by intrathecal IFNγ.19–21 In fact, there is considerable evidence that chronic pain hypersensitivity depends on NMDA receptors.30–33 It is commonly known that NMDA receptor activation is an essential step in initiating and maintaining activity-dependent central sensitization, and it critically contributes to the development of pain hypersensitivity after peripheral tissue damage or nerve injury.3,30,31,33 On the other hand, no acute changes in AMPA-induced current were observed after superfusing IFNγ into the SG neurons of rat spinal cord slices.
Tsuda et al. clearly demonstrated that the IFNγ/IFNγ receptor system is critical in activating resting spinal microglia, thereby linking them to tactile allodynia. They indicated that spinal microglia are directly stimulated by intrathecal IFNγ and contribute to IFNγ receptor-dependent pain hypersensitivity. 20 Our findings also showed that IFNγ-induced enhancement of NMDA currents was microglial IFNγ receptor-dependent. The present results are strongly supported by those of a previous study. 20
Notably, CCR2 blockade inhibited the IFNγ-induced enhancement of NMDA-induced inward currents and CCL2 increases NMDA current in SG neurons. Gao et al. 34 demonstrated electrophysiologically that CCL2 can very rapidly (within 2 min) increase NMDA currents in dorsal horn neurons. 34 They showed that incubation of the mouse spinal cord slices with 100 ng/ml CCL2 induced substantial activation of extracellular regulated kinase (ERK) in lamina I–II, and ERK phosphorylation was primarily induced in NeuN-positive neurons, indicating that CCL2 may directly act on neurons. CCL2/CCR2 signaling plays a crucial role in processing neuropathic pain.35–37 For instance, exogenous spinal administration of CCL2 induces mechanical allodynia in wildtype but not CCR2 knockout mice, 37 and in a model of neuropathic pain the development of mechanical allodynia is totally abrogated in CCR2 knockout mice. 38 These results are consistent with ours, and in addition, we found that CCL2 lies downstream of IFNγ in terms of NMDA-induced currents.
Conclusions
In summary, our results show that IFNγ enhanced NMDA-induced inward currents in SG neurons through the activation of IFNγ receptors in microglia, and CCL2 was one of the mediators between microglia and neurons. Thus, IFNγ plays an important role as one of the initiating agents of glia–neuron interactions that lead to neuropathic pain due to central sensitization.
Footnotes
Authors’ contributions
MS performed or contributed to all the experiments, analyzed data, and drafted the paper. MY and NN contributed to the experiments and analysis. ST and HY participated in study design. MY, TN, and WT contributed to experiment conception and design and edited the manuscript. All authors participated in discussion of this project and contributed to data interpretation. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by MEXT KAKENHI 26462253 to WT, MEXT KAKENHI 25460730 to TN, and MEXT KAKENHI 25462303 to ST.