
Abstract
Background
Bone metastases occur frequently in advanced breast, lung, and prostate cancer, with approximately 70% of patients affected. Pain is a major symptom of bone metastases, and current treatments may be inadequate or have unacceptable side effects. The mechanisms that drive cancer-induced bone pain are not fully understood; however, it is known that there is sensitization of both peripheral bone afferents and central spinal circuits. It is well established that the N-methyl-D-aspartate receptor plays a major role in the pathophysiology of pain hypersensitivity. Inhibition of the non-receptor tyrosine kinase Src controls N-methyl-D-aspartate receptor activity and inhibiting Src reduces the hypersensitivity associated with neuropathic and inflammatory pains. As Src is also implicated in osteoclastic bone resorption, we have investigated if inhibiting Src ameliorates cancer-induced bone pain. We have tested this hypothesis using an orally bioavailable Src inhibitor (saracatinib) in a rat model of cancer-induced bone pain.
Results
Intra-tibial injection of rat mammary cancer cells (Mammary rat metastasis tumor cells -1), but not vehicle, in rats produced hindpaw hypersensitivity to thermal and mechanical stimuli that was maximal after six days and persisted for at least 13 days postinjection. Daily oral gavage with saracatinib (20 mg/kg) beginning seven days after intra-tibial injection reversed the thermal hyperalgesia but not the mechanical allodynia. The analgesic mechanisms of saracatinib appear to be due to an effect on the nervous system as immunoblotting of L2-5 spinal segments showed that mammary rat metastasis tumor cells-1 injection induced phosphorylation of the GluN1 subunit of the N-methyl-D-aspartate receptor, indicative of receptor activation, and this was reduced by saracatinib. Additionally, histology showed no anti-tumor effect of saracatinib at any dose and no significant effect on bone preservation.
Conclusions
This is the first demonstration that Src plays a role in the development of cancer-induced bone pain and that Src inhibition represents a possible new analgesic strategy for patients with bone metastases.
Background
Cancer is the second leading cause of mortality worldwide with three of the most common cancers (breast, prostate, and lung) having a predilection to metastasize to bone, favoring the axial skeleton (vertebrae, ribs, long bones, and pelvis1–4). An estimated 70% of breast cancer patients with advanced stage disease will be diagnosed with bone metastases, which puts them at risk of skeletal-related events, including pain, hypercalcemia, spinal cord compression, decreased mobility, and fractures. It is estimated that each year in the UK, there are 30,000 patients with cancer-induced bone pain, 5 which can be severe and significantly diminish patients quality-of-life. As the tumor grows, breakthrough pain occurs and the analgesic treatments available, including opioids, non-steroidal anti-inflammatory drugs, bisphosphonates, and radiotherapy, have limited efficacy. As advances in cancer therapeutics steadily increase the life expectancy of patients, including those with bone metastases, we urgently need to develop new approaches in order to provide mechanism-based therapies to better manage cancer-induced pain.
Despite the high incidence of cancer-induced bone pain, the mechanisms that initiate and maintain it are still not completely understood. It is well known that consequences of tumor growth include tissue damage, release of inflammatory mediators, and injury to sensory nerve fiber terminals in bone, all factors that contribute to the inflammatory 6 as well as the neuropathic 7 components of cancer-induced pain. 8
Several animal models have been used to investigate bone cancer pain,9–12 in which tumor cells are injected directly into the tibia or femur of rodents, causing changes in pain-related behaviors that are thought to mimic bone pain. In animals with cancer-induced bone pain, there are changes in both the central and peripheral nervous system8,12–17 that contribute to synaptic plasticity in pain-related spinal cord neurons.18,19 This form of synaptic plasticity involves activation of G-protein coupled membrane receptors and intracellular signaling pathways that converge on the protein tyrosine kinase Src (Figure 1).
Src regulation of NMDA receptors in spinal neurons. Spinal neuron NMDA receptors are physiologically blocked at the resting potential due to a combination of Mg2+ block and dephosphorylation of protein tyrosine kinases. After intense nociceptive stimulation neuropeptides such as substance P (SP) and calcitonin gene-related peptide (CGRP) are also released from small diameter afferents in addition to glutamate. This causes activation of G-protein coupled receptors and initiates multiple intracellular signaling cascades, including phospholipase C, protein kinase C, CAKβ, and RACK1. These intracellular signaling cascades increase intracellular [Ca2+] and converge on the tyrosine kinase Src, which is phosphorylated. Src is thought to act as a hub for regulation of the NMDA receptor, and NMDA receptor subunit phosphorylation leads to increased channel gating and increased NMDA receptor activity. This increased NMDA receptor activity causes increased neuronal depolarization and sensitization (increased suprathreshold responsiveness, expanded receptive fields, sub-threshold events are amplified), leading to pain hypersensitivity.
Src is a non-receptor protein tyrosine kinase that plays a multitude of roles in cell signaling. 20 It is mainly present in osteoclasts, 21 platelets, 22 and neurons 23 and is part of the N-methyl-D-aspartate (NMDA) receptor complex23–26 in neurons. It has been widely demonstrated that increased spinal NMDA receptor activity leads to neuronal sensitization and ultimately to hypersensitivity in different pain models including bone cancer pain19,27; phosphorylation of the GluN1 and GluN2B subunits are thought to be the molecular drivers of this hypersensitivity.14,28–30 Liu et al. 31 showed that in inflammatory and neuropathic pain states, blocking the anchoring of Src with the NMDA receptor prevented the phosphorylation-mediated enhancement of NMDA receptors, resulting in reduced pain hypersensitivity. Moreover, in the same study Src−/− knockout mice showed diminished central sensitization-mediated nociceptive behaviors as well as impaired mechanical and cold allodynia in neuropathic pain. 31 As these mice had normal behavioral responses to acute noxious stimuli, Src function is thought to be crucial for pain hypersensitivity but not for normal nociceptive processing.
Src activity is not specific for pain, as both preclinical and clinical studies have reported that Src signaling is involved in other processes, including bone cell activity.21,32 Src is highly expressed in osteoclasts and controls bone resorption, and inhibiting Src activity 33 precludes osteoclast formation as well as preventing migration of osteoclast precursors with subsequent formation of actin ring and resorption pits. 34 Additionally, mice lacking the Src gene develop osteopetrosis, mainly due to impaired osteoclastic function. 35 It is interesting to note that bisphosphonates, which inhibit osteoclast function, also have some analgesic efficacy in humans, 36 and therefore, controlling bone resorption could represent an avenue for pain relief.
Despite all of these findings, the effects of Src inhibition on bone cancer pain remain to be established. We hypothesized that it may be possible to reduce cancer-induced bone pain using a Src inhibitor, which could have the novel effects of both reducing NMDA receptor activity and inhibiting bone resorption. This may represent an innovative strategy to manage bone pain in cancer patients. Here, we provide the first demonstration that Src inhibition with saracatinib reduces pain behaviors in a rat model of bone cancer pain, and that this is associated with reduced phosphorylation of spinal GluN1 receptors and reduced osteoclast activity.
Materials and methods
Experimental animals
Male Sprague Dawley rats (Harlan, 200–225 g at time of surgery) were used for the study. All animal procedures were authorized by the UK Home Office and were in accordance with International Association for the Study of Pain guidelines. Animals were assigned to groups by randomization and a group size of n = 8 was used for all studies on the basis of power calculations. The researcher performing the experiments was blind to the grouping of individual animals until the end of the experiment, although the physical appearance of saracatinib when used for gavage is such that the vehicle and lowest dose (2 mg/kg) can be distinguished from higher concentrations (5 and 20 mg/kg).
Cell preparation
Syngeneic mammary rat metastasis tumor cells-1 (MRMT-1; Riken, Japan) were cultured in Rosewell Park Memorial Institute 1640 with 10% foetal bovine serum, 1% L-glutamine, and 2% penicillin/streptomycin. On the day of surgery, cells were suspended in Hank’s buffer to achieve the final concentration of 3 × 103 cells/5 μl and kept on ice.
Cell injection
Rats were anaesthetized with 2% isoflurane in oxygen and a dental drill was used to bore a hole in the antero-medial surface of the left distal tibia. Cells were injected using a Hamilton syringe, and the hole was sealed using bone wax followed by bone cement. After irrigation with 0.9% saline, the wound was closed using sutures. The sham group underwent the same procedure but the animals received injection of Hank’s buffer instead of tumor cells. After the surgery, animals recovered after 24 h before behavioral testing.
Behavioral assessment
Prior to surgery, all animals had baseline tests of gait 37 nociceptive sensitivity to measure paw withdrawal thresholds and latency in response to mechanical 38 and heat 39 stimuli. After surgery animals were allowed to recover for 24 h and gait, paw withdrawal thresholds and latencies were measured at repeated intervals postsurgery. Behavioral testing of nociceptive sensitivity was performed on the plantar surface of the left hindpaw using calibrated von Frey filaments to measure paw withdrawal threshold, determined using the up-down method, 40 and radiant heat stimuli for measurements of paw withdrawal latency.
Bone analysis
At the end of the experiment, tibias were collected and prepared for tumor volume and bone analysis using histomorphometry. Briefly, left and right tibia were isolated, muscle removed and the bones were fixed in 4% paraformaldehyde for two days. Bones were then decalcified for three weeks in ethylenediaminetetraacetic acid, paraffin embedded, sectioned, and mounted onto slides. Total tumor area was measured on four nonserial H&E-stained histological sections and average tumor area/bone (TV/BV) calculated using Osteomeasure (Osteometrics, Decatur, GA, USA) software.
Pharmacokinetics
Whole blood was obtained via cardiac puncture under terminal pentobarbital (200 mg/kg i.p.) anaesthesia and centrifuged (4℃ for 5 min at 11,000 rpm) to separate the plasma, which was frozen at −80℃ until use. Saracatinib levels were measured by liquid chromatography-mass spectrometry (Charles River, Tranent, UK) using a API5000 mass spectrometer (AB Sciex, Ontario, Canada). The lower limit of the assay was 0.5 ng/ml and the upper limit was 5,000 ng/ml.
Western blotting
Proteins were extracted from the dorsal L2-5 segments of the spinal cord in lysis buffer (RIPA buffer plus phosphatase inhibitor; PhosSTOP, Roche) with an ultrasonicator on ice, and cleared of cellular debris and nuclei by centrifugation at 14,000 relative centrifugal force for 15 min at 4℃. Extracted proteins (25 µg per well) were loaded and separated by standard 3% to 8% Tris-Acetate gel. Proteins were transferred to polyvinylidene difluoride membranes and then blocked with 5% (w/v) dried milk in TBS-T for 1 h at room temperature. The blots were incubated overnight at 4℃ using the following antibodies that were diluted in TBS-T containing 5% dried milk: phosphorylated GluN1 (Ser896) 1:10,000 (Millipore, cat # ABN88); GluN1 1:1,000 (Millipore cat# AB9864R); phosphorylated Src (Tyr418) 1:800 (Abcam, cat# ab4816); Src 1:800 (Abcam, cat# ab47405). The following day, the primary antibody was bound with a goat anti-rabbit secondary antibody that was conjugated to horseradish peroxidase (Cell signaling, cat #7074-S) and the signal detected by enhanced chemiluminescence. Each phosphoprotein was normalized to the expression of the corresponding total protein on the same membrane. Densitometric analyses were performed using Image J software.
Bone biomarker analysis
Under terminal pentobarbital (200 mg/kg, i.p.) anesthesia, whole blood was obtained by cardiac puncture and centrifuged (4℃, 2000 rpm for 15 min), TRAP5b and procollagen type 1 N-terminal propeptide (P1NP) were measured by ELISA (Immuno diagnostic systems, ELISA kit for TRAP5b cat# SB-TR102 and PNP1 cat# AC-33F1) using 100 µl samples of serum according to the manufacturer instructions.
Compounds
Saracatinib was provided by AstraZeneca. The doses chosen were based on previous rat studies where saracatinib was shown to inhibit Src kinase in tumor tissue in vivo. Pharmacokinetic studies have shown that there is some central nervous sytem (CNS) penetration with saracatinib in rats with cerebrospinal fluid (CSF) levels being about 3% of plasma levels. The vehicle for saracatinib was a mixture of 0.5% hydroxypropyl methyl cellulose (Fluka) + 0.1% polysorbate (Tween 80; Fluka), sterilized by autoclaving at 121℃ for 20 min at 30 psi (1.5 Bar).
Statistical analysis and data presentation
Data are shown as means and the standard error of the mean (± SEM) of three rats (for Western blotting) or eight animals (for in vivo behavioral studies). Statistical evaluation was performed by Mann Whitney test and one- or two-way analysis of variance (ANOVA), followed by appropriate post hoc tests. The a priori level of significance at 95% confidence level was considered at p < .05.
Results
Saracatinib inhibits thermal hyperalgesia in a rat model of bone cancer pain
We used a rat model of bone cancer pain to study the effect of Src inhibition on gait, paw withdrawal latency to heat stimuli, and paw withdrawal thresholds to mechanical stimulation. Rats received intra-tibia injection of mammary cancer cells MRMT-1 (3 × 103 in 5 µL) and gait analysis as well as behavioral responses to mildly noxious mechanical and thermal stimuli applied to the hindpaw were determined prior to and at different time points post-injections. Compared to Hanks buffer-injected controls, there was no significant change in either the area of the affected paw that was used during locomotion, or the pressure applied to the paw during the step cycle in animals that had intra-tibial MRMT-1 cell injections (data not shown). In our hands, flinching and guarding were not reproducible findings in MRMT-1 treated rats, and there was no qualitative difference between control and treated animals. Nonetheless, MRMT-1 injected rats showed time-dependent, significant (p < .05, two-way ANOVA) reductions in hindpaw withdrawal latency to thermal stimuli and in mechanical withdrawal threshold relative to the Hank’s buffer-treated controls (Figure 2 (a) and (b)). Signs of hypersensitivity were detectable as early as Day 1 post-MRMT-1 injection, were well established by Day 6 and persisted to at least Day 13 postsurgery.
Pain-related behavior after intra-tibia MRMT-1 injection. (a) Mean (±1 SEM) paw withdrawal latency to radiant heat stimulation of the ipsilateral plantar hindpaw in animals injected with either MRMT-1 cells or vehicle in the left tibia (n = 8/group). The MRMT-1 injection was performed on day 0 (indicated by the dashed line). Asterisks indicate significant differences (p < 0.05; 2-factor repeated-measures ANOVA followed by Tukey’s test post hoc) between the MRMT-1 and vehicle injected rats. (b) Mean (±1 SEM) 50% paw withdrawal thresholds to graded mechanical stimulation in the same animals as shown in (a). Asterisks indicate significant differences between MRMT-1 and vehicle injected animals (p < 0.05; 2-factor repeated-measures ANOVA followed by Tukey’s test post hoc).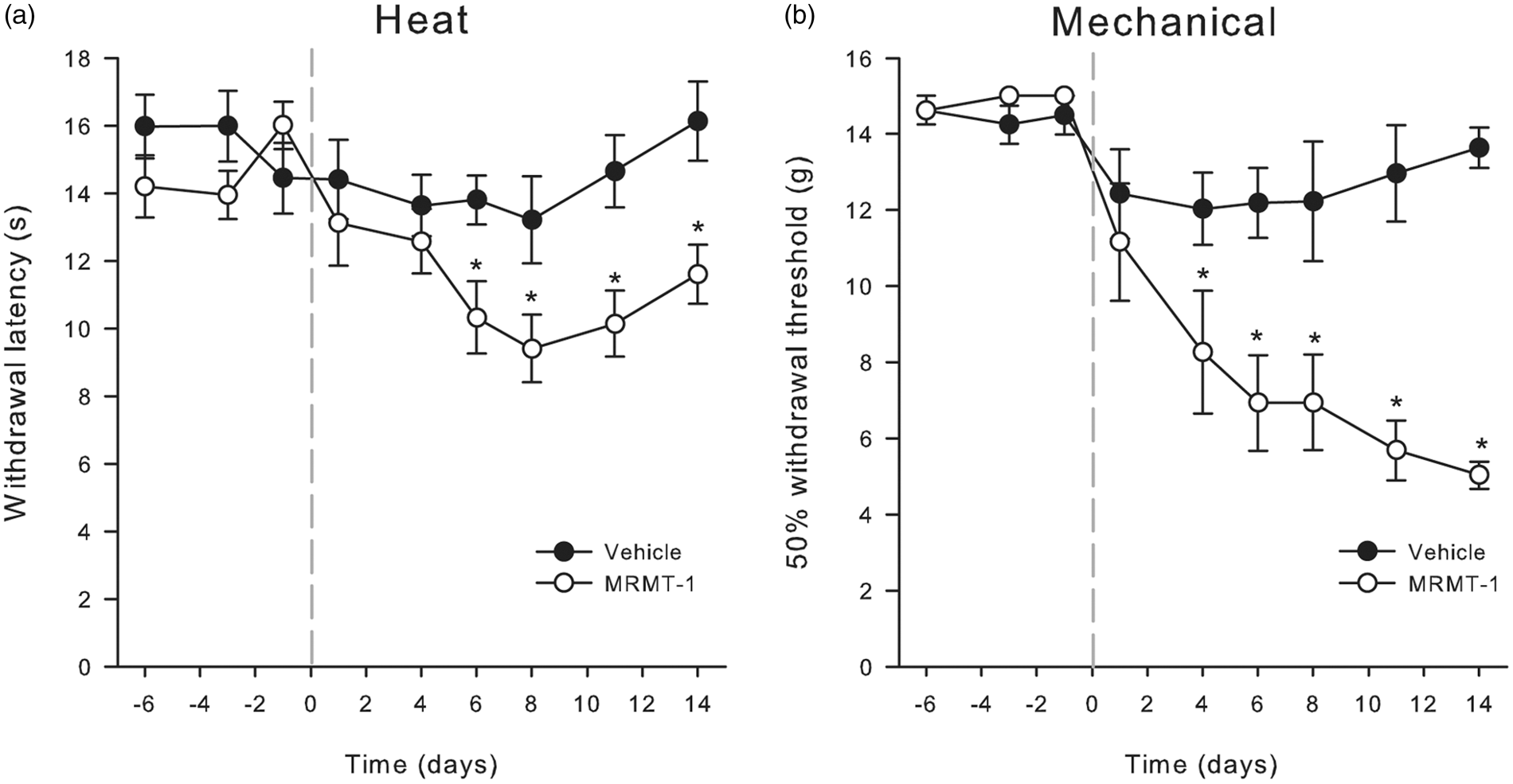
In order to examine the effect of Src inhibition on the MRMT-1-induced hypersensitivity, rats were treated daily with saracatinib by gavage for seven days (either 2, 5, or 20 mg/kg, n = 8/group), beginning seven days after MRMT-1 or Hank’s buffer injection. Saracatinib is an orally bio-available, small molecule that inhibits Src and Bcr-Abl.
41
Saracatinib (20 mg/kg, p.o. for seven days) fully reversed cancer-induced heat hyperalgesia (Figure 3(a)), but lower doses were ineffective (data not shown). Daily injection of saracatinib (20 mg/kg but not 2 or 5 mg/kg) in MRMT-1 tumor-bearing rats resulted in withdrawal latencies that were significantly (p < .05, two-way ANOVA) elevated relative to those measured in MRMT-1/vehicle-treated rats (Figure 3(a)). None of the doses of saracatinib used were able to reduce tactile allodynia (Figure 3(b)). Administration of saracatinib (20 mg/kg, p.o.) to Hank’s buffer-treated animals did not produce any change in either paw withdrawal latency or paw withdrawal threshold (Figure 3(a) and (b)), indicating that saracatinib does not inhibit normal nociceptive processing.
Reversal of heat hyperalgesia by Src inhibition in cancer-induced bone pain. Daily gavage with saracatinib (20 mg/kg) reversed the heat hyperalgesia (a), but not the mechanical allodynia (b) of cancer-induced bone pain. Symbols show mean ± 1 SEM paw withdrawal latencies to radiant heat stimulation (a) or 50% withdrawal thresholds to graded mechanical stimulation (b). Animals (n = 8/group) received an intra-tibial injection of either MRMT-1 cells or Hanks buffer vehicle on day 0 (gray line) followed by daily administration of either saracatinib or vehicle starting on day 7 (black line). Lower doses of saracatinib (2 and 5 mg/kg) were ineffective (data not shown). Asterisks indicate significant differences between MRMT-1 injected animals treated with either saracatinib or vehicle (p < 0.05; 2-factor repeated-measures ANOVA followed by Tukey’s test post hoc).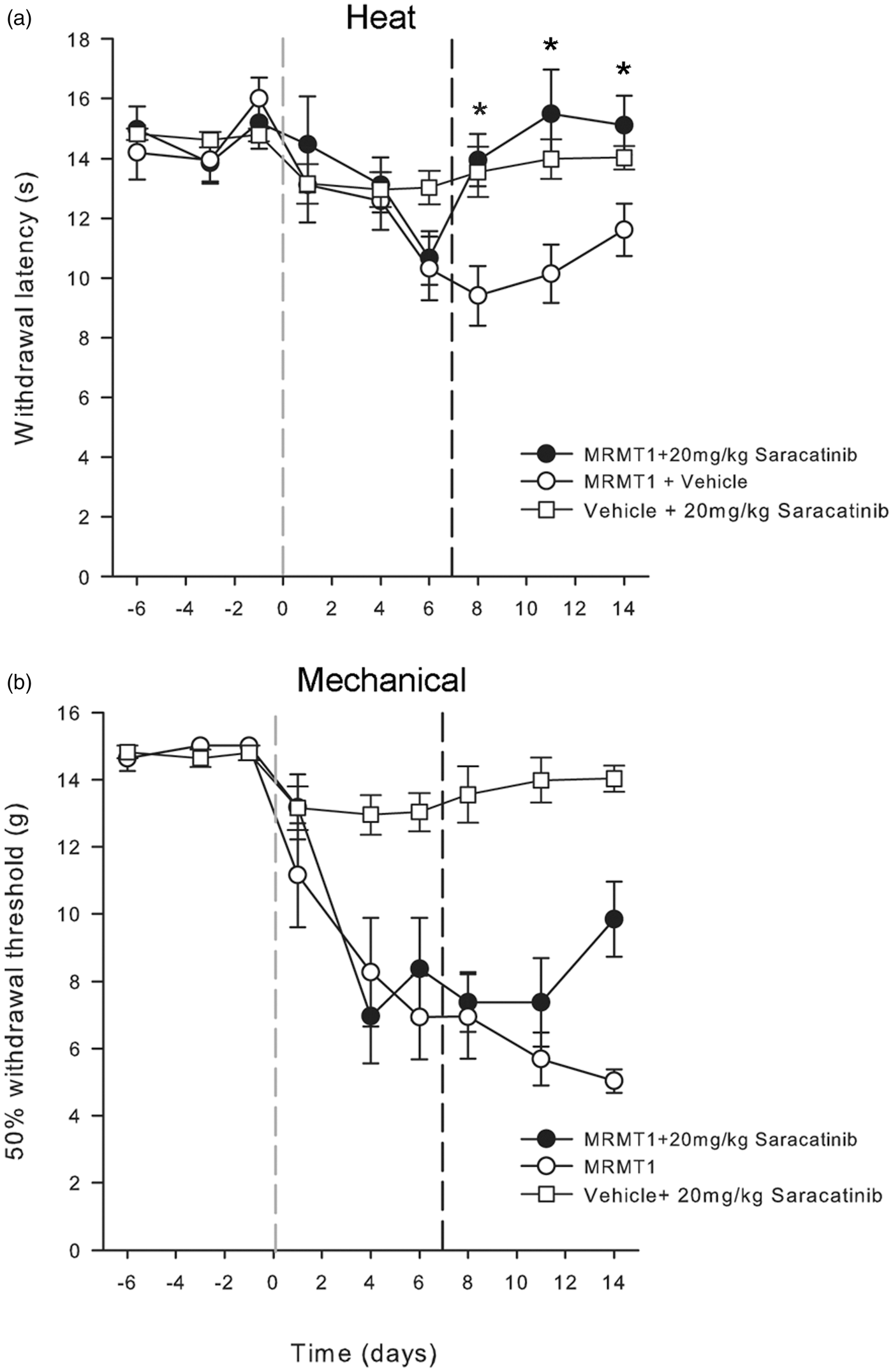
Increasing the dosing duration of animals treated with 20 mg/kg of saracatinib from 7 to 11 days had no effect on tactile allodynia (p > .7, two-way ANOVA, data not shown). Pharmacokinetic studies of different saracatinib doses and exposure times (measured from plasma samples taken at the end of the dosing period) showed a nonlinear relationship between plasma saracatinib concentration and dose, and in the 20 mg/kg group increasing the dosing duration from 7 to 11 days did not produce any additional increase in plasma saracatinib levels (p > .05; one-way ANOVA; Figure (4)). Saracatinib has good oral bioavailability and a t1/2 of approximately 7 h in rats; plasma levels taken 24 h after 7 or 11 days daily dosing at 20 mg/kg demonstrated free levels of saracatinib > 50 × Ki for Src kinase (Ki 2.7 nM). In addition, from previous studies in rats, the CSF exposure of saracatinib is 3% of total plasma levels which would provide compound exposure in the CNS of at least 10 × Ki for Src at trough. Hence, based on the known pharmacokinetics in rats, plasma protein binding and the measured saracatinib exposure data in these studies, levels of saracatinib are expected to inhibit Src kinase in peripheral tissues and the CNS by 100% for the 24-h period after daily oral dosing. Therefore, its failure to reduce mechanical allodynia is unlikely to be due simply to sub-optimal administration, in terms of either frequency or dose.
Pharmacokinetics of Saracatinib in vivo. Box plots of plasma saracatinib concentration measured by high-performance liquid chromatography following dosing at 2, 5, or 20 mg/kg daily for 7 days and at 20 mg/kg for 11 days. The horizontal line within the box is the median, the box boundaries are the 25th and 75th percentiles, and the error bars show the data range. There is a nonlinear relationship between saracatinib dose and plasma levels but extending the treatment duration from 7 to 11 days at 20 mg/kg did not increase the plasma concentration of the drug (p > 0.05, ANOVA).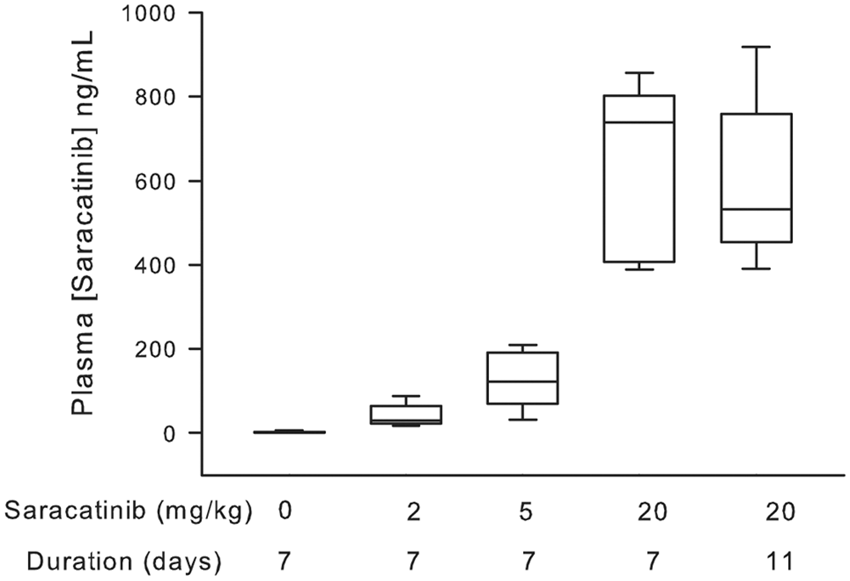
Taken together, these studies showed that inhibiting Src selectively abolishes heat hyperalgesia in a model of cancer-induced bone pain but does not affect normal nociceptive processing. Tactile allodynia was unaffected by Src inhibition, even when the dosing period was extended, suggesting different underlying mechanisms in the pathophysiology of thermal hyperalgesia versus tactile allodynia in bone cancer pain.
Saracatinib reduces spinal GluN1 receptor phosphorylation in cancer-induced bone pain
Saracatinib could have its effect on behavior in cancer-induced bone pain by an action on nociceptive circuits within the nervous system or by preserving bone due to reduced osteoclastic resorption. Src is widely expressed in the nervous system, being enriched in the cerebral cortex, hippocampus, brainstem, cerebellum, and spinal cord.22,42 The effect(s) of saracatinib could potentially occur at several levels within the nervous system, and the current study was not designed to elucidate its specific site of action. Instead, we have investigated whether MRMT-1 cell injection modulated Src activity and/or downstream signaling pathways in the Src cascade in the nervous system, and if treatment with saracatinib could modulate this activity. Because the first synapse in nociceptive pathways is found in the spinal dorsal horn, we studied Src and GluN1 activation in the spinal cord.
MRMT-1 cells or Hank’s buffer were injected into the left tibia and seven days later rats were treated either with saracatinib (20 mg/kg, p.o.) or vehicle (1 ml/kg, p.o.) by daily gavage. In a terminal experiment, the dorsal horn from the left L2-5 segments of the spinal cord was harvested at either 1, 6, 8, 11, or 13 days postintratibial injection for immunoblotting analysis of phosphorylation of Src and GluN1, an NMDA receptor subunit that is downstream of Src (Figure 5(a) and (b)). About 40% of the afferents that innervate the tibia originate from the L3 dorsal root ganglion with the remainder being scattered across the L2–L5 ganglia.
43
Most of the spinal neurons activated by acute noxious stimulation of the tibia are in the L3 and L4 spinal segments
44
; therefore, to ensure that as much of the innervation of the tibia was analyzed, we used L2–L5 spinal segments in the blotting experiments.
Saracatinib inhibits GluN1 phosphorylation in spinal dorsal horn. Injection of MRMT-1 into the left tibia of rats resulted in increased phosphorylation at the spinal level of the nonreceptor tyrosin kinase Src (a) and of the NMDA receptor subunit GluN1 (b), as determined by Western blotting and band densitometry, β-actin was used as an additional loading control (c). Saracatinib (20 mg/kg/day, p.o.) treatment significantly (p < 0.05, Mann-Whitney test) reversed phosphorylation of Src (a) in a time-dependent manner and significantly (p < 0.05, Mann-Whitney test) reduced phosphorylation of GluN1 within 24 h from the first injection (b). Blot label: C = control; d = day; v = vehicle; s = saracatinib.
Intra-tibial injection of MRTM-1 cancer cells, but not Hank’s buffer, resulted in a significant (p < .05, Mann Whitney test) increase in Src phosphorylation that was detectable one day postinjection and remained elevated up to the last time point studied, Day 13 (Figure 5(a)). Similarly, phosphorylation of the GluN1 subunit was significantly increased (p < .05, Mann Whitney test) in MRMT-1 treated rats from Day 1 to at least Day 13 (Figure 5(b)). Daily treatment with saracatinib (20 mg/kg, p.o.), but not vehicle, in MRMT-1 tumor bearing rats significantly (p < .05) reduced phosphorylated Src to levels comparable to that observed in Hank’s buffer-injected rats (Figure 5(a)). Expression of phosphorylated Src was reduced (59% change compared to MRMT-1 treated rats) within 24 h of the first saracatinib dose (Day 8 post intra-tibial injection), and this effect was maintained throughout the full administration period (observable on Days 11 and 13 post-intra-tibia injection, 52% and 87% change compared to MRMT-1 treated rat, respectively; Figure 5(a)). Importantly, saracatinib also significantly decreased phosphorylation of the GluN1 subunit of the NMDA receptor (Figure 5(b)). Similar to the inhibition of MRMT-1-induced Src phosphorylation with 20 mg/kg saracatinib daily, phosphorylation of GluN1 was reduced within one day of the first dose and this persisted throughout the duration of the study (Figure 5(b)).
Here, we demonstrate that animals with behavioral signs of cancer-induced bone pain show increased activation (phosphorylation) of Src and the GluN1 subunit of the NMDA receptor in segments of the lumbar spinal cord that are known to receive primary afferents from the tibia.17,43,44 We confirm with immunoblotting that saracatinib-inhibited spinal Src in vivo, as it reduced MRMT-1 induced Src phosphorylation, and importantly, saracatinib also reduced GluN1 phosphorylation in the spinal dorsal horn whilst also reversing heat hyperalgesia behavior at the whole animal level. Overall, this suggests that the Src-inhibitor saracatinib has a major effect on the nervous system and, therefore, potential efficacy as an analgesic in cancer-induced bone pain.
Src inhibition has minimal effects on inhibiting bone resorption
In order to establish whether the effects of saracatinib on behavior were in part due to the bone-preserving effects of Src-inhibition, we investigated bone histology and biomarkers of bone turnover. The left tibia of rats was inoculated with MRMT-1 cells or Hanks’ buffer and the animals treated by daily gavage with saracatinib (2, 5, or 20 mg/kg) seven days later for a further seven days. At the end of the experiment, tibias were collected and processed for bone histomorphometry to determine effects on tumor and bone volume/integrity. Saracatinib, including the 20 mg/kg dose known to reduce pain-related behaviors, did not have any effect on tumor growth in the tibia (Figure (6)). Additionally, analysis of bone turnover biomarkers showed that saracatinib dose-dependently reduced serum levels of tartrate-resistant acid phosphatase tartarate-resistant acid phosphatise (TRACP5b), an enzyme highly expressed by bone resorbing osteoclasts (Figure 7(a)). Seven days of saracatinib administration (5 or 20 mg/kg, p.o. daily) significantly (p < .05, one-way ANOVA) reduced serum levels of TRACP5b in tumor-bearing rats compared to tumor-bearing rats that received vehicle (Figure 7(a)). Saracatinib at 2 mg/kg, p.o. had no significant effect (p = .22, one-way ANOVA; Figure 7(a)). No significant difference was observed in serum levels of P1NP (a marker of osteoblast activity) in tumor bearing rats following treatment with saracatinib (2, 5, or 20 mg/kg, p.o. per seven days) when compared to vehicle treated rats (p = .49, p = .52, and p = .71, respectively; one-way ANOVA; Figure 7(b)). These data show that whilst saracatinib at a dose that reverses heat hyperalgesia behaviorally does reduce bone resorption, it has no significant effect on bone formation, suggesting that the antiresorptive effect of saracatinib has only a minor role in its efficacy as a potential analgesic.
Saracatinib has no effect on MRMT-1 tumor growth. Photomicrographs of haematoxylin and eosin (H&E) stained decalcified sections of tibia injected with either vehicle (a) or MRMT-1 cells (b, c). Low power images of whole bones (ai, bi, ci) show the normal marrow in the control group and the extent of tumor growth in the MRMT-1 injected groups 13 days after injection (arrowheads in bi and ci). As can be seen in (c), saracatinib 20 mg/kg p.o. for seven days had no effect on tumor cell growth. Higher magnification images show the drill hole (aii, bii, cii) and the marrow spaces (aiii) and the tumor-bone interface (biii,ciii). Quantitative analysis of multiple sections through the tibia showed that saracatinib had no significant effect on tumor growth (d) at any dose tested. Bars show mean + 1 SD with n = 3/group. Effects of Src inhibition on bone turnover. Treatment with saracatinib for seven days (2, 5, or 20 mg/kg/day, p.o.) significantly (p < 0.05, one-way ANOVA) reduced the serum levels of TRACP-5b (a) in a dose dependent manner. However, daily injection of saracatinib did not change the serum levels of PNP1 (b).

Discussion
The present study demonstrates that Src inhibition has the potential to play a role in reducing cancer-induced bone pain. Intratibial injection of MRMT-1 cancer cells has previously been shown to cause thermal hyperalgesia and mechanical allodynia of the skin,10,17 and here we show that inhibiting Src reverses MRMT-1 induced cutaneous heat hyperalgesia, probably due to an action on the nervous system. Additionally, we showed that the analgesic effect of saracatinib is not due to inhibition of tumor growth, and it is also independent of its effects on bone.
Technical considerations
The current study used behavioral testing of the plantar hindpaw skin to infer the presence of bone pain. Cancer patients with bone metastases report altered skin sensation overlying sites of skeletal disease when compared to control sites 45 ; the majority of patients reported increased skin sensitivity in response to pinprick or warm stimuli, but brushing and cool hyperesthesia were also reported and the heat hyperalgesia and mechanical allodynia observed in the current study is consistent with this. In cancer patients, there was no attempt to relate the proximity/area of abnormal skin sensation to the location/size of the skeletal metastasis, 45 the skin sensory changes can cover up 140 cm2, 45 indicating that cancer-induced bone pain evokes a zone of secondary hyperalgesia in the overlying skin that can spread over a considerable area, and this could account for why tumor cells growing in the rat distal tibia cause behavioral changes to plantar skin stimulation. Many previous animal studies of cancer-induced bone pain have used the surrogate measure of behavioral changes to mildly noxious cutaneous stimuli to infer the presence of bone pain, with a few studies assessing noncutaneous behavioral changes such as weight-bearing, grip strength, and rotarod tolerance. Use of a Hank’s buffer control shows that the behavioral changes to cutaneous stimulation are not simply a postsurgical effect, and we, therefore, assume that they reflect the effects of the tumor cells within the tibia. It is interesting to note that of the 31 studies of cancer-induced bone pain in the literature where MRMT-1 cells were used in rats, 46 28 studies used cutaneous stimuli applied to the plantar foot as a surrogate measure for bone pain, 2 did not report any behavioral testing, and only 1 used deep tissue stimulation (mechanical pressure on the antero-medial surface of the distal tibia 47 ) but there was no comparative plantar skin testing in that study.
Role of Src in the nervous system in cancer-induced bone pain
In rodents it is known that cancer-associated pain is associated with increased phosphorylation of Src, GluN1, and GluN2B in spinal cord, and that these changes are thought to contribute to the generation and maintenance of pain.30,48,49 We confirmed that intra-tibial MRMT-1 injection evokes phosphorylation of spinal GluN1 receptors using an antibody to the S896 site of the GluN1 receptor; importantly, we show that GluN1 phosphorylation is inhibited by Saracatinib at a dose that reverses heat hyperalgesia. We interpret the increased GluN1 phosphorylation as receptor activation and conclude that Saracatinib reduces GluN1 activation by inhibiting Src, but the precise relationship between S896 phosphorylation and pain is unknown. Because Src is a molecular point of convergence through which a variety of intracellular signaling cascade and cell-surface receptors enhance NMDA receptor function, its activation has been implicated in the synaptic plasticity that is required for physiological processes such as learning and memory as well as pathological events like persistent pain.23–26,31 Long-term potentiation (LTP) plays a key role in long-lasting increases in synaptic transmission and one of the major mechanisms of LTP is by the actions of NMDA receptors on ion currents. It has been shown that altering Src functioning by intracellular administration of Src blockers inhibits LTP induction. 50 Changes in synaptic transmission as a result of NMDA receptor upregulation is also crucial in the maladaptive plasticity associated with pathological conditions such as persistent pain. Woolf and Salter 19 have described how spinal dorsal horn nociceptive neurons are hyper-responsive in several pain models as a result of enhanced NMDA receptor activity, which is critical for the initiation and maintenance of increased neuronal responsiveness. Thus, the analgesic effect of saracatinib might be due to inhibition of the direct activation/phosphorylation of the GluN1 NMDA receptor subunit.
Consistent with previous reports, 29 we have demonstrated that spinal levels of phosphorylated Src and GluN1 are elevated in rats with tumor in bone compared to sham animals. Inhibiting GluN1 phosphorylation with either a protein kinase A inhibitor 51 or antisense oligodeoxynucleotides 52 reduced some of the behavioral signs of neuropathic and inflammatory pain in earlier studies. Although GluN1 phosphorylation was reduced by Saracatinib in the present study and, therefore, may be important in cancer associated bone pain, other targets downstream from Src may also be involved. We attempted quantitative immunoblotting of the phosphorylated form of the GluN2B subunit of the NMDA receptor using a Tyr1472-phospho GluN2B antibody and by immunoprecipitation using a pan-phosphorylated antibody followed by a GluN2B subunit antibody. GluN2B is implicated in the mechanisms of chronic pain as preventing its phosphorylation reverses inflammatory pain hypersensitivity 30 and blocking spinal GluN2B subunits reduced hyperalgesia/allodynia in the MRMT-1 model of cancer-induced bone pain. 14 Despite previous results with the Tyr1472-phospho GluN2B antibody, 14 we were unable to demonstrate reproducible high-quality immunoblotting of phosphorylated GluN2B and immunoprecipitation was also unsuccessful.
Previous studies have shown that altering Src activity affects behavioral responses to different modalities of noxious stimuli equally: heat and mechanical hyperalgesia in a model of inflammatory pain were both reduced in mice when Src was decoupled pharmacologically from the NMDA receptor and Src−/− knockout mice show reduced hyperalgesia to both mechanical and cold stimuli in a model of neuropathic pain. 31 However, in the current study, Saracatinib only reversed heat hyperalgesia, having no effect on mechanical allodynia. We assume that there is a causal relationship between inhibition of Src/GluN1 phosphorylation and reversal of heat hyperalgesia by Saracatinib in treated MRMT-1 rats, but current models of central sensitization would predict that when postsynaptic NMDA receptor activity is reduced, hypersensitivity to all types of noxious stimuli would be affected. 19 One speculative explanation for the observation that Saracatinib did not ameliorate mechanical allodynia relates to Src expression in functionally specific groups of nociceptive neurons. It is an unwritten assumption that Src is expressed equally in all neurons at the same anatomic location, but which might have different functional properties. This is probably an incorrect generalization on the basis of Src immunocytochemical labeling in the cortex, where it is clear that even though there is strong staining of layer V pyramidal neurons, not all neurons are labeled. 22 There are no immunocytochemical studies of Src in the spinal cord, but there is no a priori reason to assume that all nociceptive spinal neurons express Src equally. It is known from previous in vivo studies of spinothalamic and spinoparabrachial neurons that functionally specific subgroups of nociceptive neurons exist.53,54 While the most frequently encountered nociceptive cell types respond to several different stimulus modalities (heat, cold, mechanical, etc.), a subset selectively responsive to noxious mechanical stimuli is well documented. 53 Perhaps, the neurons that receive just noxious mechanical inputs express less Src than those that receive both mechanical and heat inputs and, therefore, are less likely to be inhibited by Saracatinib. This could be tested in future anatomical studies.
Src inhibition and bone turnover
Previous studies in mice lacking the c-Src proto-oncogene have shown that Src expression is necessary for normal bone resorption, as these animals develop osteopetrosis, 35 and the most critical role for c-Src in bone is related to osteoclast function rather than differentiation. 55 There is no difference in the number of formed osteoclasts between wild type and Src−/− knockout mice; however, in Src−/− knockout mice, osteoclasts failed to form ruffled borders, resulting in a limited ability to resorb bone matrix. 55 In vitro studies suggested that Src also regulates the life span of the osteoclast, thus affecting bone resorption.56,57 In contrast however, Src−/− knockout mice showed equal or higher numbers of osteoclasts than wildtype mice, suggesting that in vivo there is a compensating mechanism from other members of the Src kinase family. 35 In accordance with these previous reports showing that Src inhibition influences the activity of osteoclasts and not their numbers, the data from our study demonstrate that saracatinib, in a dose dependent manner, reduced the level of the systemic bone resorption marker TRACP5b, similar to the effects observed in patients with advanced cancer. 58 Clinical dose escalation studies with saracatinib have shown that doses of 50 to 175 mg/day, which are well tolerated in humans with solid tumors, produce a reduction in bone resorption markers including TRAP5b without altering tumor burden.55,58 Therefore, these data suggest that Src kinase inhibition reduces bone turnover in both species and acts as a biomarker of Src inhibition in these studies. In contrast with other reports in which disruption of Src activity enhanced osteoblast differentiation and bone-forming activity, 59 in our study saracatinib treatment had no effect on P1NP serum levels of tumor-bearing rats. Although saracatinib reduced osteoclast activity, this finding did not translate to bone preservation, potentially due to the very aggressive nature of the model with high numbers of osteoclasts at the tumor-bone interface. It has been demonstrated that saracatinib reduces tumor growth in a c-Src-transfected 3T3-fibroblast xenograft model in vivo 41 ; however, in our study, seven days of saracatinib treatment failed to influence the tumor growth in the tibia at any of the doses tested, further suggesting that the antihyperalgesic effect observed of saracatinib is independent of tumor volume, and it is mainly mediated by its effects on the nervous system.
Conclusion
These findings demonstrate a role for Src in bone cancer pain and indicate that inhibition of Src through saracatinib may represent a novel strategy for reducing bone cancer pain. Critically, the effects of saracatinib on pain behaviors seem mainly due to its effects on the nervous system, by inhibiting phosphorylation of Src and GluN1, and not due to an antitumor or bone preserving effect of the drug. This suggests that a better understanding of how NMDA receptors subunits are implicated in development and maintenance of bone cancer pain may lead to more mechanism-specific therapeutic strategies.
Footnotes
Acknowledgement
We are grateful for the expert technical assistance provided by Cai Price, Diane V Lefley, and Holly Evans.
Authors’ Contributions
DA, IH, DWL, JE, and MDF conceived the study and designed the experiments, MDF performed experiments, MDF and DA analyzed data, MDF and DA wrote the manuscript. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: MDF, DWL, IH, and DA declare that they have no competing interests. JE is an employee of Astra Zeneca.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by funds from the United Kingdom Medical Research Council under the “Mechanisms of Disease” initiative.