
Abstract
Canada has high rates of generic usage, but the mechanisms of ensuring timely generic entry are dysfunctional. Unlike most other countries with a linkage regime in which regulatory approval depends on clearing patent hurdles, Canada does not offer temporary generic exclusivity. Instead, it grants the generic kept out of the market by a patent that is ultimately found invalid or not infringed the opportunity to seek its lost profits from the patentee. The system is unbalanced in that patentees find it profitable to litigate even very weak patents since they sustain a monopoly; while the incentives for generic firms to litigate, even if they expect to win, are very weak. This paper analyzes the incentives of the parties, and proposes possible policy fixes to help rebalance the system.
Introduction
The Patented Medicines (Notice of Compliance) Regulations (“PM(NOC) Regulations”) govern the process for market entry of new medicines in Canada. 1 In Canada, the regulations operate as a “linkage” regime, tying regulatory approval of generic or biosimilar medicines (i.e., medicines for which regulatory approval is sought on the basis of a comparison with, or reference to, an already approved drug) to the protection of patent rights. The asserted goal of the regulations is “to balance effective patent enforcement over new and innovative drugs with the timely market entry of their lower-priced generic competitors.” 2 The regulations prior to 2017 created a system with two possible steps for patentees to limit generic competition: first, they could bring an application to enforce patents through the linkage system, and then, if unsuccessful, they could begin an action to sue the generic for patent infringement. Reforms enacted in 2017 replaced this risk for dual litigation within an action to determine patent validity and infringement.
The Regulatory Impact Analysis Statement stated that the amendments would resolve numerous problems, including eliminating the “costly and inefficient practice of dual litigation.” 2 Because the single action includes discovery and live witness testimony, patentees and generic firms can advance more evidence. In addition, the amendments removed barriers to litigating patents “outside the Regulations” prior to generic entry.
It is reasonable to ask, with several years of experience, whether the amended regulations are indeed balancing patent enforcement with timely generic market entry. Achieving this delicate equipoise is not only challenging but is equally difficult to measure. Ideally, the regulations should provide consistency so that both patentees and potential generic entrants can reasonably predict the outcome of actions, and thus avoid the cost of an action. Moreover, the regulations should make it unattractive for the patentee to litigate a patent that would, if litigated, be found invalid or not infringed; and for a generic firm to assert invalidity or non-infringement for a patent that would, if litigated, be found valid and infringed.
The incentives created by the regulations do not align with these ideals. The central problem is that there are strong incentives for patentees to assert and litigate patents even if they expect to lose; and weak incentives for generic firms to litigate patents even if they expect to win. This arises from the imbalance in the rewards to maintaining a monopoly versus the rewards to entering a competitive market. It is not difficult to see why this imbalance arises: even if a patent is found invalid or not infringed, the brand firm selling a product for a high price earns much more from its continued sales than it needs to pay out in damages to a generic litigant kept out of the market. At the same time, the benefit to the generic firm from engaging in litigation is limited by competition. I return to this discussion below, with specific examples.
By way of background, I note that generic medicines are a key component of the Canadian healthcare system. Generic drugs make up 75% of all prescriptions filled in Canada but account for only 22% of drug spending. 3 The use of generic drugs is estimated to save Canadians $37 billion annually. 4 While most provincial health budgets are extremely strained, generic drugs represent a critically important source of budgetary relief. This makes it crucial to ensure that the regulations governing the entry of generic drugs into the market are well designed to achieve entry as soon as possible while respecting the rights properly accorded to patentees.
In the analysis that follows, I examine how the regulations have changed outcomes and how they are working. I begin, however, by considering how the market has changed in other ways in the past 10 years.
Background on the generic drug industry
A generic drug is a prescription pharmaceutical product which is sold as being bioequivalent to a brand, “innovator” or reference pharmaceutical product. 1 Bioequivalence implies that the generic drug can be expected to have the same therapeutic effects and safety profile as the reference product when administered to patients under the conditions specified in the approved labelling. When qualified as bioequivalent by Health Canada, generic drugs are functionally substitutable for one another and for the brand drug. 5 Provinces have interchangeability rules that authorize or mandate pharmacists to substitute a lower-priced generic drug for the branded product (unless the prescription specifies no substitutions).6–8
Generic drugs typically are lower-priced than the brand-name equivalent because the generic supplier competes on price, while the brand-name equivalent drug is priced to take advantage of its monopoly power. Typically, generic drugs dominate markets given their price advantage and often the brand-name drug exits the market in the years following generic entry. Thanks to the lower price point of generic drugs, they offer very large savings to patients and insurers.
Biosimilar drugs are “highly similar” to a biologic drug already authorized for sale. Biosimilars differ from generic drugs in that the reference product is not considered to be bioequivalent, and as a result pharmacists cannot automatically substitute a biosimilar for the reference biologic drug. However, there are generally no clinically meaningful differences in efficacy and safety between a biosimilar and the reference product. In Canada, the provincial insurance plans increasingly require that new patients are initiated on the biosimilar, and that existing patients are transitioned to the biosimilar, given a lower price. The model for marketing biosimilar drugs is different from that of generic drugs, although a lower price for a very similar product remains the key competitive advantage and benefit for payers.
The generic drug industry in Canada consists of over 100 companies that supply generic drugs, including numerous large companies that operate internationally. Some manufacture drugs in Canada, although most import drugs from suppliers abroad. Some firms specialize in a therapeutic area or type of product. Some have an in-licensing model, in which they import finished drugs from abroad and sell them in Canada, having undertaken the necessary regulatory submissions. The Ontario formulary, which lists most drugs available in the province, illustrates the range of firms: for example, as of November 2024, Apotex, Canada’s largest generic drug firm, has 712 products listed in the formulary; Altius has 3. 9
As with brand name drugs, production has been globalized. While some firms have domestic production – much of which is exported – the generic drugs that Canadians use are most often imported. 10 Economies of scale in production make it efficient to produce each drug in a limited number of facilities. Globalization is most apparent for biosimilars, whose complex manufacturing processes have led to relatively few global manufacturing plants. Thus, for biosimilars, it is common for there to be in-licensing rather than domestic production.
Generic drug pricing is highly regulated in Canada and has steadily declined over the past 20 years. The Patented Medicine Prices Review Board (PMPRB) has tracked nominal prices for generic medicines, which do not include off-invoice discounts companies granted to pharmacies. It found that wholesale prices for generic drugs had fallen by 59% between 2007 and 2018. 11 Additional reductions have pushed generic drug prices even lower. The PMPRB analysis captures manufacturer prices only, excluding off-invoice discounts, and thus does not reflect the net prices received by manufacturers. (These discounts reduce the prices paid by pharmacies.) This makes international comparisons based on manufacturer prices into an apples-to-oranges comparison; a more helpful analysis is one that compares actual prices, and the only level at which actual prices are available is the retail level. A 2023 report found that the average Canadian retail price to the consumer was approximately one third lower than the average foreign price after accounting for distribution fees and mark-ups. 2
In summary, the Canadian generic industry is highly competitive and faces aggressively regulated pricing. Generic firms in Canada do not operate on large margins. This is relevant for understanding the incentives of generic firms to engage in litigation over patents that they believe may be found invalid or not infringed.
The assessment of how the 2017 amendments to the PM(NOC) Regulations affected the balance between supporting innovation and enabling timely access to low-priced generic drugs is complicated by two important changes in the industry that also occurred in the same year. Under a Supreme Court decision in 2017, the “Promise Doctrine” was eliminated. 12 The Promise Doctrine had put a patent’s validity at risk “if even one ‘promised’ use [was] not soundly predicted or demonstrated.” Eliminating the Promise Doctrine shifted the balance of protection significantly towards the patentee. Also in 2017, Canada implemented a new Certificates of Supplementary protection regime, as required under a trade agreement between Canada and the European Union. This measure provided an additional period of protection for drugs containing a new medicinal ingredient, or a new combination of medicinal ingredients, protected by an eligible patent. It is important to keep these two developments in mind when interpreting data regarding how litigation has changed in Canada since the PM(NOC) amendments were implemented in 2017.
It is also important to recognize that generic entry in Canada typically occurs, in cases with litigation, long before the last asserted patent expires. The first generic NOC in litigated cases (regardless of the outcome) precedes the expiration of the last listed patent on the Patent Register by about 48 months on average. This indicates that litigation continues to be necessary; if generic firms were to wait for all patents on the Register to expire, the additional expense to Canadians could be considerable. Thus, there are significant risks to public budgets from failing to get the “balance” right: extending the average duration of brand name drug exclusivity by 48 months would cost provincial health plans, private insurers, and Canadian patients billions of dollars a year.
Assessing the regulatory balance
The duration of exclusivity
One way of assessing the balance between generic entry and the protection of patentee interests is the length of exclusivity. Using data on court cases in Canada and the Notice of Compliance database, I was able to identify that the average length of exclusivity for the brand name drugs that faced litigation increased following the Amendments from 12 years 9 months (4663 days) to 12 years 11 months (4723 days). While this sounds relatively minor, it is financially impactful if sustained. Annual spending on patented medicines was $18.4bn in 2022. If the average generic drug is priced at 30% of its patented equivalent, then the additional annual cost of increasing the average exclusivity period by 1 day is (1/4723) × $18.4bn × (100% - 30%) = $2.7 m. Thus, the additional annual expense of increasing the exclusivity period by 60 days is $163.6 m, or approximately 1% of annual spending on patented prescription drugs.
It is also helpful to compare this to the United States. I identified the set of drugs that have faced litigation under the new PM(NOC) regulations for which there has been generic entry in both Canada and the US.
3
I calculated the difference between the dates of first generic Notice of Compliance in Canada and the first approval of an Abbreviated New Drug Application for a generic in the US.
4
Canadian approvals were, on average 152 days, or about 5 months, later than in the US. The difference is shown on the horizontal axis of the Figure 1 below. The brand revenues in the year prior to generic entry are shown on the vertical axis. The size of each circle is scaled to reflect the impact of the number of years difference in approval dates times the brand revenues. In effect, the circle size can be seen as the additional savings achievable if Canada were to have had the US approval date (on the left-hand side) or the additional cost from having the Canadian instead of the US approval date (on the right-hand side). Brand revenues and generic approval times in the US and Canada. Notes: The horizontal axis shows the number of days difference in approval time of the first generic for each product by the US FDA and Health Canada. The vertical axis shows the annual sales of the brand product in the calendar year prior to generic entry in Canada. The size of the bubbles is scaled to represent the additional cost (on the left) or savings (on the right) had Health Canada approved the generic drug at the same time as the FDA.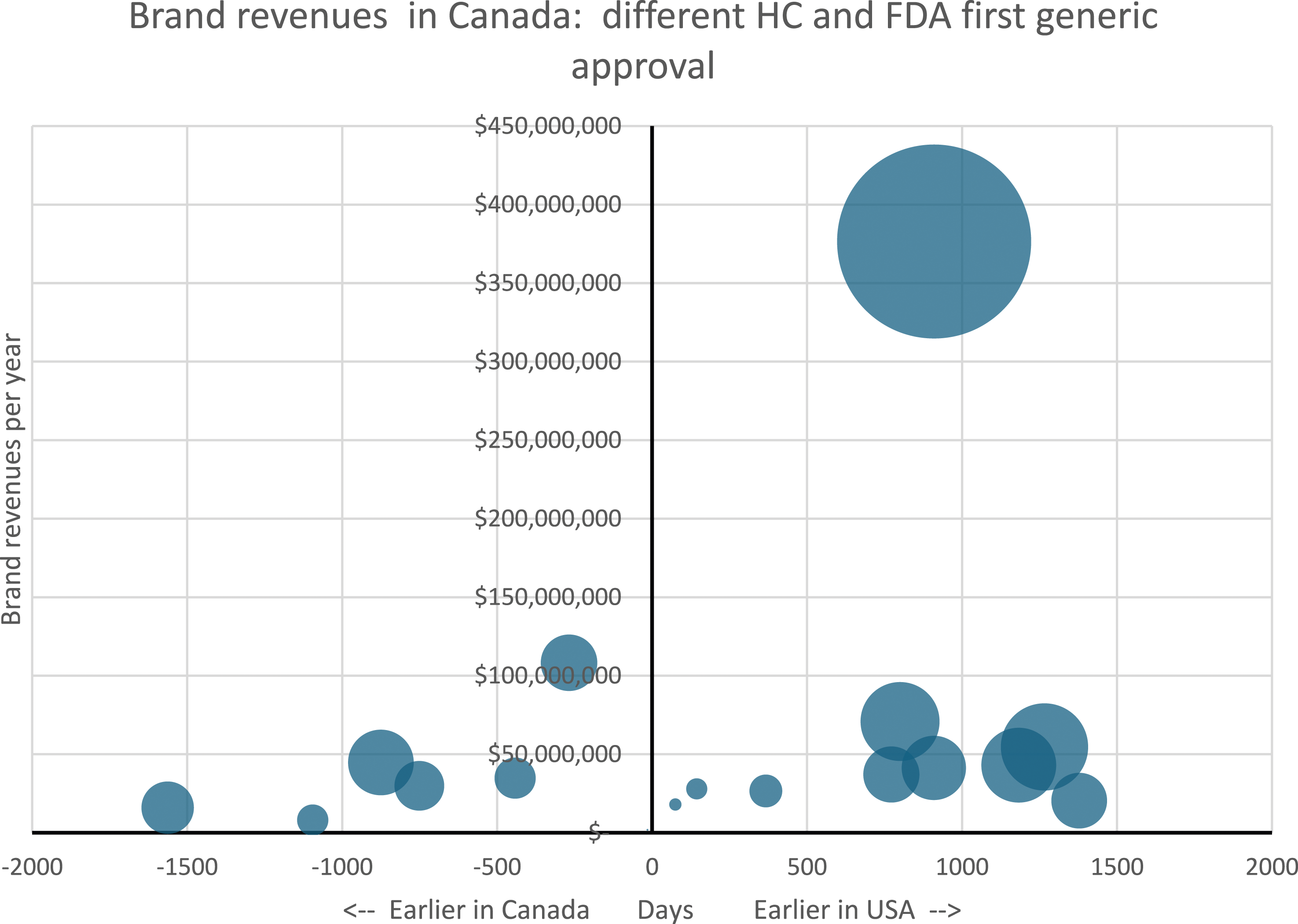
One should be careful in over-interpreting these results, as the data includes only products for which a generic or biosimilar equivalent has been approved in both Canada and the US. In addition, the set of products is relatively small, and regulatory approval is not the same thing as market entry. However, for at least this set of drugs for which generic entry has proceeded under the post-2017 regulations, regulatory approval for generics in Canada has been relatively slow on average.
Number and type of patents
There has been a steady increase in the number of patents litigated per drug, with the average standing at around 2.5 since 2017, compared to about 2.0 in the previous 12 years. This increase in the number of patents is largely due to an increase in the number of “use” patents, which now feature in almost 40% of cases, compared to only 24% in the previous 12 years. Since patent applications occur long before patents are litigated, we cannot attribute the change in patent numbers or their type to the amendments in the PM(NOC) Regulations. However, these features of the patent landscape are important background. With the increase in the number of patents, and the recent challenges created by jurisprudence around “skinny labelling”, generic firms are facing increasing costs and risks of entry. 5
The costs of litigating
As discussed above, one of the goals of the Amendments was to reduce the costs of dual litigation, which arose in some cases, particularly those with high value to the brand. However, since all litigation under the PM(NOC) Regulations now requires a full action, with all the costs of discovery and live witnesses, the costs of every case have now gone up. It is difficult to obtain clear data on these costs.
I interviewed external and internal counsel for several generic drug manufacturers. A consistent observation was that the costs of a full action are considerably higher than those of an application under the old regulations. Typically, the costs are estimated to be in the range of $2 m–$6 m, depending on the complexity of the case and the number of patents. Costs appear to have roughly doubled, compared to those of a simple application under the old regulations. Moreover, some cases are advancing on a dual track as patents not registered in the Health Canada registry may be asserted under S. 8.2 of the new regulations. In these cases, litigation costs are even higher.
If a firm carries its case, it is generally granted a fraction of a costs, which must be paid by the opposing party. I heard from my interviewees that the fraction of costs covered was typically around 30%. I also heard that patentees tend to have costs on the upper end of the range.
A generic company considering litigation must therefore count the costs carefully. If it “wins” it can be expected to have net court costs in the range of $1.4 m–$4.2 m (i.e., 30% less than the gross costs. If it “loses”, it will have to pay its own costs plus 30% of the patentee’s costs, perhaps $3 m–$8 m in total. These costs appear to be a strong disincentive for generic firms to engage in litigation. 6
If a generic firm “wins” in an NOC action, it may be entitled to seek damages under Section 8 of the regulations, based on the loss caused by market entry delay because the patentee asserted infringement. Determining the amount of the loss, however, is complex, since it depends on numerous counterfactuals, such as what market share the generic firm would have obtained, what price it would have charged, what discounts it would have offered, and what other firms would have done, all in the hypothetical circumstances of earlier entry. Estimating these counterfactuals is complex and subject to dispute, and therefore requires a whole new round of costly litigation. A trial for such damages could be expected to cost millions of dollars.
The value of “winning”
If a generic and brand firm persist in litigating a case and the patentee wins, it continues to enjoy its monopoly profits: 100% market share and a high price. If it loses, at least it has maximally extended its exclusivity period. In either case, litigating is highly attractive for patentees for all but the smallest markets.
The incentives to litigate are much less attractive for generic firms. If the generic firm enters the market based on patent invalidity, other generics may also soon enter. 7 If the generic firm enters based on non-infringement, other generic firms with a non-infringing technology may in some cases also be able enter. The patentee’s interests in keeping out subsequent entrants typically ends after one has entered, since the patentee will typically rapidly lose market share regardless of the number of generics. According to interviews with counsel for generic companies, the terms of settlements with generic firms often give them the right to enter at the same time as or shortly after the first generic. In some cases, the generic litigant may be able to enter earlier than other generic firms, which gives it a competitive advantage. In other cases, the litigating generic will earn no more than generic firms that settled, since all will enter simultaneously. The effect is that generic firms’ incentives to litigate are very weak, since there is no intrinsic market advantage created by litigation; non-litigating firms may benefit equally without bearing any costs.
The product pirfenidone presents the model of how the system is working for generic manufacturers. 8 Generic and biosimilar drugmaker Sandoz sought, under the regulations, to bring a generic version of the branded drug Esbriet. After a lengthy process, and then a trial, it was successful and immediately brought a product to market, having obtained an NOC on 2021-05-03. Other generic firms, however, had been watching, but do not seem to have been investing heavily in litigation. After settling with the patentee, JAMP and Teva obtained NOCs on 2021-05-18, just 2 weeks later. Under Canadian pricing regulations, the price of pirfenidone fell to 50% of the brand price. The generic firms will have offered off-invoice discounts to pharmacies in order to earn their business. I do not have specific information about costs or discounts in this case, but if the cost of manufacturing was 20% of the brand price; and off-invoice discounts offered by Sandoz and JAMP were 20% of their price, or 10% of the brand price, then Sandoz’s pirfenidone would have earned only 20% of the brand price in profits per unit (=50% - 20% - 10%), while the brand would have been earning 80% (=100% - 20%). The brand earned revenues of roughly $38 m in 2020. Data protection for the brand product expired on 2020-10-01. So, a delay of generic competition for 7 months would be very attractive and would have enabled the brand to capture profits of roughly 7/12 × $38 m × 80%, or $17 m. Even if it had to pay damages and court costs, the brand would want to litigate even if it was certain to lose.
Depending on the exact circumstances, Sandoz may be able to claim damages for being kept out of the market. Its damages would however be limited to its expected profits. And assuming that Sandoz would have obtained, say, a market share of 40%, with JAMP obtaining a market share of 40% and the brand name product taking the other 20%, Sandoz’s damages would be only around 7/12 × $38 m × 20% x 40% = $1.7 m. Given expected costs of over a million dollars in the Section 8 damages case alone, $1.7 m would hardly make it worth engaging in litigation, even if its legal costs were partly compensated by the patentee. Looked at from the perspective of challenging the patent in the first place, the benefit of challenging can only dominate the risks of substantial costs if the generic firm believes it has a very high probability of winning at trial.
Generic litigation does not always lead to early entry. If a generic firm litigates and loses at trial, it faces considerable risks: for example, Apotex attempted to supply a generic paliperidone palmitate product. Apotex sought a summary judgement in order, presumably, to limit costs. This was, however, not entirely successful, since both Apotex and the plaintiff, Janssen, retained several experts and had several lawyers represent them in court for 3 days. At trial it was determined that Apotex’s product would induce infringement. 13 Apotex therefore had to pay its own costs in addition to those of the patentee. In a case that went to a full trial, Pharmascience attempted to supply a generic sitagliptin product. Seven experts testified and nine lawyers are listed as appearing in the case, which lasted for 10 days in court. The trial transcript is over 1300 pages. We can anticipate that the costs of litigation were several million dollars. Pharmascience was unsuccessful and it was determined that its generic product would infringe Merck’s patents. 14 Pharmascience will have had to bear very substantial costs.
These cases nicely summarize why there is a problem in the incentives for generic firms to invest in litigation. It is almost always better to settle with the brand in order to be assured of being allowed to enter at the same time as the first generic, since that avoids court costs and the risks associated with litigation. As a result, even if everything goes well for a generic that chooses to litigate, the outcome is typically a competitive market of limited profitability – other generic firms are likely to be waiting to enter as soon as the litigating firm succeeds. The risks are similar for small-market drugs (such as some orphan drugs), even where a second generic entrant seems improbable, since the court costs and the probability of losing at trial can overwhelm any potential profits from earlier entry. And the risk of losing is that the generic manufacturer is barred from entry and has to pay very substantial legal costs.
One approach to addressing the risks inherent in going to trial is for the patentee and the generic to settle on a date for entry. The balance of incentives, however, clearly favours the brand: the generic has little to fight for given that the patentee can settle with the non-litigating generic competitors on the basis of simultaneous entry. Section 8 of the PM(NOC) Regulations is supposed to provide an incentive for generic litigants, since they can be compensated for lost sales. However, patentees can substantially diminish the calculated damages by settling with multiple generic firms to allow them entry at the same time as the first generic. The strategy of settlement is dominant for small generic firms, and in many circumstances even for large generic firms, and it is easy for the patentee to offer such settlements, which reduce the incentive for any firm to engage in litigation. In effect, for generic firms, the upside of litigating is very small, and the downside can be quite large. There have, as a result, been very few Section 8 cases filed recently. 9
Summary
The discussion above shows that the current PM(NOC) regulations result in weak incentives for generic firms to litigate, even though there is enormous social value from weak patents being challenged. It is profitable for a patentee to litigate over a patent even if it is highly likely to lose at trial. And it can be unprofitable for a generic to litigate even if it is highly likely to win. I also note that there has been an increase in the period of exclusivity – i.e., a delay in generic entry – after 2017. In addition, regulatory approval of generic drugs that are litigated appears to be relatively slow, compared to the US.
Correcting the balance
Canada is facing unprecedented challenges in funding its health care systems, so it is essential to strike the right balance in terms of supporting innovation through the patent system while not delaying generic competition undeservedly. This section discusses possible policy fixes to resolve the current weak incentives for litigation by generic firms. The proposals below differ in their complexity and difficulty of application; some involve relatively minor tweaks to the current regulations, and others would involve an overhaul.
Temporary generic exclusivity
The approach taken by the US the Hatch-Waxman Act is that a successful generic challenger is rewarded with 180 days of quasi-exclusivity, before any other generic firms are approved to enter. 10 While creating incentives for litigation by generics, this approach has also attracted considerable criticism. 15 Early problems in the operation of the 180-day exclusivity provisions in the Hatch Waxman Act led it to have a reputation as being problematic, although after reforms enacted in 2003, the exclusivity provisions appear to be working well and delivering effective incentives to challenge patents.
South Korea has had, since 2012, a “linkage” system like that in Canada and the United States in that regulatory permission to market a generic drug is linked to the patent system. In South Korea, the patent linkage system grants the first generic drug to make a successful patent challenge a 9-month period of exclusivity. The exclusivity award is considered as an important factor in ensuring generic entry. 16
Taiwan has used a linkage system since 2019. The regulatory authority stays approval of a generic drug for 12 months if the patentee files a patent infringement suit. If the generic is found not infringing or the patent expires or is found invalid, the first generic applicant is granted a 12-month exclusivity period, starting on the date the product is marketed. 17
A similar system operates in China, where a generic that invalidates relevant patents, thus opening the door to generic competition, is eligible for a 12-month exclusivity period. 18 Notably, the Chinese system requires invalidation, so that a generic drug that avoids infringement by designing around patents does not qualify for the exclusivity award. A generic drug first earned this exclusivity only in 2024, for Chia Tai Tian Qing’s everolimus product. 19
Among countries with linkage regimes, Canada is now an outlier in not offering an exclusivity reward for the first filing generic. The adoption of the generic exclusivity provision in Asian countries, modeled on the US’s Hatch Waxman Act, shows a recognition that, when regulatory approval and patent status are linked, some further incentive is required to induce generic companies to invest in litigation, beyond merely being able to enter the market and compete against companies that did not invest.
Damages to payers
Australia has taken a completely different tack; rather than trying to increase incentives for generic firms to challenge weak patents, it tried to increase the cost to patentees of litigating weak patents. Specifically, it has attempted to incorporate the government as a potential damages claimant when a patentee has delayed generic entry and then lost at trial. 20 Such an approach, were it to be applied in Canada, would make it much more costly for a patentee to litigate a weak patent, since there would be a risk of having to pay out damages to the generic firm and to payers who been denied the opportunity to buy the generic product. There are numerous challenges to implementing such a scheme. Most notably, the obvious strategic solution for a patentee and generic challenger would be to settle on terms favourable to the challenger, since patentees would want to remove the risk of a large damages award to governments. This would not necessarily lead to earlier entry. In addition, in Canada it would not be straightforward to determine which payer would be eligible to claim the damages award, given that there are numerous provincial insurance plans and thousands of private insurance plans. Moreover, it is notable that this approach does not itself enhance the incentives of generic firms to invest in patent challenges, unless they are able to earn payments through attractive settlements with patentees.
Enhanced damages to generics excluded from the market
Another alternative would be to increase the size of Section 8 damages awards, which tend to be small for several reasons. The lost profits of the generic firms are always smaller than the monopoly profits of the patentee because the generic margin of price less cost is always much lower than that of the brand, and because a single generic firm’s share of the market is lower than 100%. Moreover, the patentee can strategically reduce hypothetical damages by, for example, settling for same-day entry with multiple firms in advance of generic entry. Damages awards could be increased to make it less attractive for patentees to litigate, and more attractive for generic firms to litigate, when patents are weak. There are several possibilities: a. Section 8 damages could include the option for an accounting of profits earned by the brand during the time that a generic was excluded from the market. This would make it much less attractive for a patentee to sue based on a weak patent and would give generic firms strong incentives to challenge weak patents. b. Damages could be based on the challenging generic’s counterfactual profits, assuming no other generic firm in the market. This would result in much higher damages than basing damages on a counterfactual with multiple competing generic firms. c. A very limited reform would be to set damages as the challenging generic’s lost counterfactual profits, assuming no generic entry on the basis of settlements undertaken between the patentee and other potential entrants. This would, at least, mean that patentees could not proactively reduce their damages exposure through settlements that did not accelerate the date of generic entry.
Simplifying damages to generic firms
A partial solution would be to simplify and clarify the calculation of damages awards under Section 8. As noted above, the complexity of calculating damages is high when the court needs to make decisions about counterfactuals. Simplifying damages calculations would have numerous advantages: (1) less court time required for Section 8 cases; (2) lower costs of litigation for both parties; (3) increased certainty of quantum of payment; and (4) less delay in payment of damages. Various possible approaches could be used. a) Damages could be calculated as the difference between the brand and counterfactual generic price times the volume of the brand for the duration of the time that a challenging generic was excluded from the market. b) A similar model could include modifications to the above. For example, the regulations could stipulate that the payment to the generic would be half the brand’s revenue for the product for the period of exclusion, with the amount to be divided between generics depending on their hypothetical potential entry date. c) Combined with one of the above, the payment of damages could also be mandated to be completed within a fixed period (e.g., 24 months).
Reducing trial costs
Finally, trial costs themselves are, as shown above, a substantial barrier for generic firms to pursuing litigation. Because patentees have incentives to litigate even weak patents, it may be attractive to increase the costs of doing so. For example, it would be reasonable to impose a 100% cost award for a patent that was dropped before trial. 11
Similarly, it may be possible to increase access to expedited mechanisms not requiring a full trial when the generic entrant clearly does not infringe the claimed patent. Such a mechanism could be applied subject to a bright line test, such as whether the generic consists of a different polymorph or is seeking to enter with a skinny label for a use that would not infringe the claimed patent. 12
Conclusion
The incentives operating in Canada’s PM(NOC) Regulations are evidently not balanced. Patentees have strong incentives to litigate even weak or clearly not infringed patents, and potential generic entrants have weak incentives to litigate even when they expect to win. This must result in patents that would be found invalid or not infringed if litigated being used to delay generic entry. The rights of patentees are fully protected by the current regulations, since no generic entry can occur without a full trial. But trials should not be used as a cudgel to delay or prevent competition, which is the inevitable result of the current regime. Adjusting the incentives through changes to the damages awards in Section 8 or creating a generic exclusivity period could help to bring the regulations back into balance.
Footnotes
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The author has been retained as an expert witness by numerous pharmaceutical companies, including generic drug companies.
Funding
The author disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant from the Canadian Generic Pharmaceutical Association.