
Abstract
Numerous successful gene-targeted therapies are arising for the treatment of a variety of rare diseases. At the same time, current treatment options for neurofibromatosis 1 and schwannomatosis are limited and do not directly address loss of gene/protein function. In addition, treatments have mostly focused on symptomatic tumors, but have failed to address multisystem involvement in these conditions. Gene-targeted therapies hold promise to address these limitations. However, despite intense interest over decades, multiple preclinical and clinical issues need to be resolved before they become a reality. The optimal approaches to gene-, mRNA-, or protein restoration and to delivery to the appropriate cell types remain elusive. Preclinical models that recapitulate manifestations of neurofibromatosis 1 and schwannomatosis need to be refined. The development of validated assays for measuring neurofibromin and merlin activity in animal and human tissues will be critical for early-stage trials, as will the selection of appropriate patients, based on their individual genotypes and risk/benefit balance. Once the safety of gene-targeted therapy for symptomatic tumors has been established, the possibility of addressing a wide range of symptoms, including non-tumor manifestations, should be explored. As preclinical efforts are underway, it will be essential to educate both clinicians and those affected by neurofibromatosis 1/schwannomatosis about the risks and benefits of gene-targeted therapy for these conditions.
Introduction
Neurofibromatosis 1 (NF1) and schwannomatosis (SWN), including NF2-SWN, LZTR1-SWN, and SMARCB1-SWN, are neurogenetic syndromes which predispose affected individuals to a range of tumors and non-tumor manifestations (Figure 1).1–3 Although the majority of these tumors are histopathologically benign, they can cause significant morbidity and mortality due to their number, size, location, and nervous system involvement.4–8 In addition, the diverse non-tumor manifestations can substantially contribute to disability and further reduce the quality of life.9–11 In this article, we summarize the main challenges of gene-targeted therapies for NF1 and SWN and discuss pathways forward to apply these technologies in this patient population.
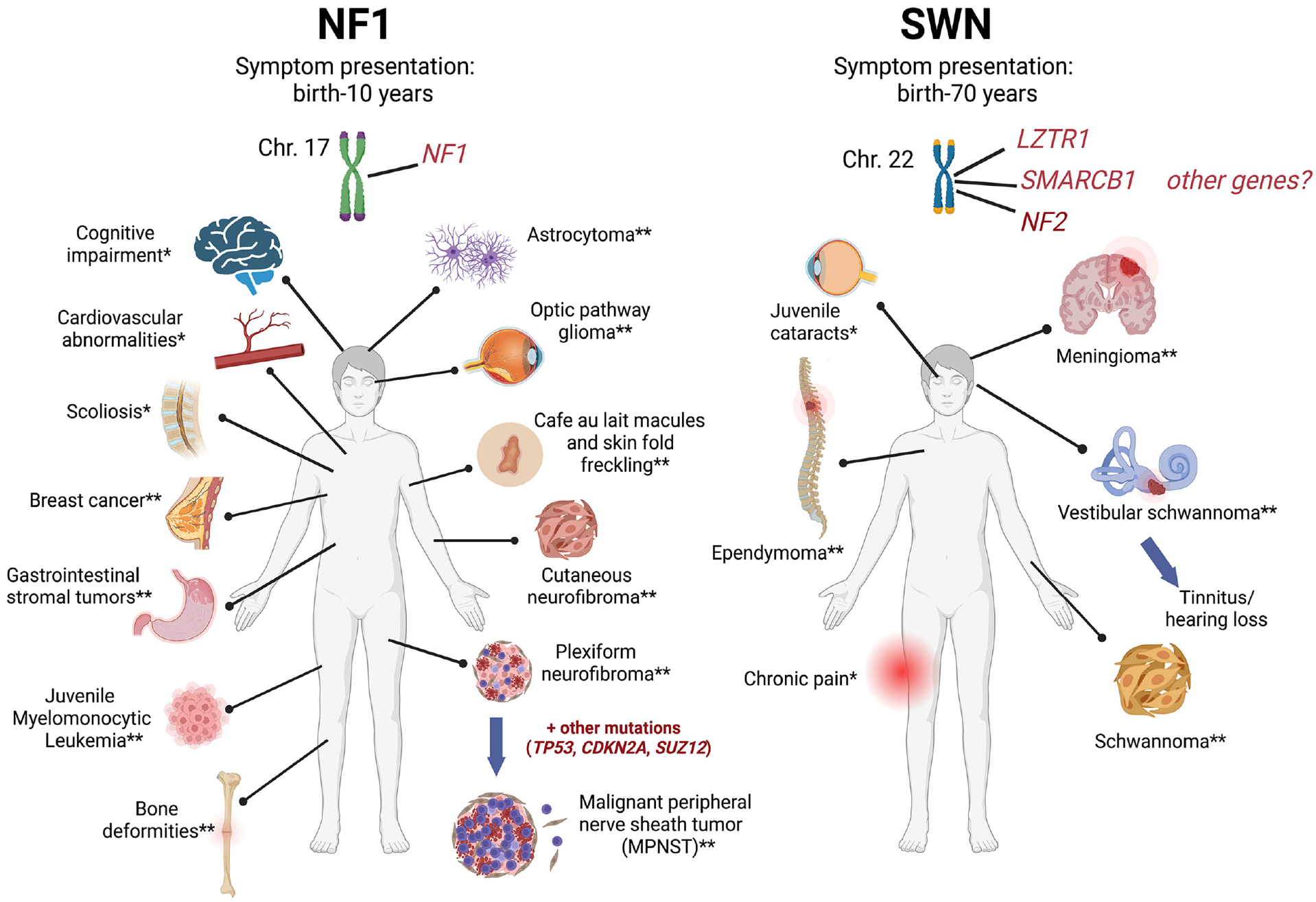
NF1/SWN symptom presentation.
Current success of approved gene-targeted therapy
Gene-targeted therapy, the process of manipulating a pathogenic gene variant or its product through a variety of different modalities, has provided novel treatment options for several inherited and acquired conditions, including hematologic disorders, immunodeficiencies, cancer, infectious diseases, ophthalmologic and dermatologic conditions, and neurological diseases. 12 As of December 2022, more than 27 gene therapy products are approved by the US Food and Drug Administration (FDA), with 5 therapies targeting monogenic disorders. 13 While these therapies may be life-changing for affected individuals, 12 they also fundamentally change how life-threatening disorders can be treated.14,15 Gene-targeted therapy is under intense investigation in oncology; however, its application for genetic conditions linked to tumor predisposition such as NF1 and SWN is still in its infancy.
Current treatment approach to NF1 and SWN
Most treatments for NF1 and SWN focus on symptomatic tumors. While surgery is the primary treatment, several chemotherapy trials for NF1 and SWN have been completed. Currently, the only drug approved specifically for people with NF1 is the Mitogen-activated protein kinase kinase (MEK) inhibitor selumetinib.6,16,17 Bevacizumab, a neutralizing antibody for vascular endothelial growth factor, is recommended for treatment of hearing loss related to progressive vestibular schwannomas in NF2-SWN. 18 Unfortunately, no clinical trials have shown a complete imaging response in NF-related tumors or complete reversal of symptoms.6,16 Current treatments also fail to address multisystem involvement of these conditions, particularly in NF1, 17 and therefore do not alleviate many important concerns of patients. 19 Moreover, current therapies do not treat the underlying genetic cause of these manifestations, thereby failing to prevent future morbidities.16,17,20 Gene-targeted therapy offers the potential to bridge some of these gaps and provide more comprehensive and efficacious treatments than are currently available.
Rationale and challenges for gene-targeted therapy for NF1 and SWN
For NF1 and SWN, gene-targeted therapies are designed to increase levels of functional proteins in cells that have either one functional gene copy, known as haploinsufficient cells, or no functional copies, defined as diploinsufficient cells, of a tumor suppressor gene. While diploinsufficiency is required for tumor formation, haploinsufficiency results in abnormal cell function that can also cause disease manifestations or can promote tumor progression through changes in the microenvironment. Thus, increasing functional protein levels holds promise for treating established tumors, for preventing tumor formation, and for treating non-tumor manifestations of these conditions.
NF1 is caused by pathogenic variants (PVs) in the NF1 gene encoding neurofibromin. 3 Several studies have demonstrated that gene transfer of a functional copy of the NF1 GRD to NF1-deficient malignant peripheral nerve sheath tumor (MPNST), glioma, and plexiform neurofibroma cells can suppress RAS-pERK signaling and inhibit growth.21–23 However, despite many years of research, gene-targeted therapy for NF1 still presents many challenges. First, NF1 has a highly variable inter- and intrafamilial phenotypic expression, likely related to family-specific NF1 PVs, unknown epigenetic, environmental, and stochastic factors, as well as involvement of multiple organ systems.3,24 Loss of heterozygosity for NF1 and resulting diploinsufficiency is required for manifestations such as tumors and pseudoarthrosis,25,26 while haploinsufficiency can drive tumors 27 and cause other phenotypes, such as neurocognitive problems (Figure 1). In addition, structural abnormalities such as those involving the bone and brain may not be reversible after a defined developmental timepoint. Second, neurofibromin expression varies between different cell types and tissues, and tight regulation of transgene expression is necessary to avoid alterations in RAS-MAPK signaling and cellular fates, resulting in potentially serious complications. Variable tissue distribution also involves unique neurofibromin isoforms, which are produced by alternative splicing. 28 The most common isoforms 1 (2818 aa) and 2 (2839 aa) differ in a 21-aa insertion (exon 30alt31, formerly 23a) within the GTPase-activated protein (GAP)-related domain (GRD), which results in a significantly reduced GTPase activity in isoform 2.29,30 Notably, deletion of isoform 2 or altering the ratio of isoform 1 to 2 may lead to learning impairments in mice or disturbed neuronal differentiation, respectively.31,32 Finally, it remains unknown how much neurofibromin must be restored and by which time point to significantly modify the cellular fate or condition.
The main challenge in treating NF2-SWN patients is that the precise functions of merlin, the NF2 gene product, are still not fully understood. Merlin has multiple roles in cell development and tumorigenesis, 33 preventing direct targeting of upstream or downstream NF2 cellular signaling pathways and thus pointing to strategies directly targeting the NF2 gene. In other molecular subtypes of SWN, individuals harbor germline PVs in SMARCB1 or LZTR1 (or in other genes yet unknown), but their role in schwannoma tumorigenesis has yet to be established. Most syndromic schwannomas, whether attributed to germline PVs in NF2, SMARCB1, or LZTR1, carry biallelic inactivation of the NF2 gene suggesting it is necessary for Schwann cell tumorigenesis.34,35 Thus, tumors in SMARCB1-, LZTR1-, and NF2-SWN patients could all benefit from NF2 gene replacement therapy, with the caveat that each schwannoma of individuals with non-NF2-SWN carries a unique somatic NF2 PV. 36 In addition, patients also present with non-tumor manifestations such as cataracts in NF2-SWN and neuropathic pain in SMARCB1- and LZTR1-SWN, and the curative effect of merlin re-expression in the affected tissues is uncertain.
Similar to NF1, diverse cell types give rise to schwannomas, meningiomas, and ependymomas; this necessitates defining the optimal target cells for treatment to avoid unnecessary exposure of off-target tissues and to optimize treatment window. The cellular responses to merlin overexpression in normal (merlin-expressing) tissues are not known. Moreover, people with NF2-SWN are merlin/NF2 hemizygous whereas people with SMARCB1- or LZTR1 PVs have normal levels of merlin throughout their body except in the tumor.
Currently investigated gene-targeted treatment modalities in NF1 and SWN
There are numerous potential gene-targeted therapy approaches and strategies that have recently been thoroughly reviewed and discussed in detail.37,38 Hence, we provide a brief overview. Understanding how each approach works informs selection of the most appropriate therapy for specific PVs, tissues, and manifestations. Genome editing can be used to alter the nucleotide sequence or the chemistry of targeted DNA bases. 39 These changes occur on a base-by-base basis, and are highly personalized, with new reagents designed for nearly each type of PV. Alternatively, additional copies of DNA (encoding all or a significant portion of a gene) can be introduced in gene replacement therapy, which either restores gene function (for most loss-of-function variants), or as suicide gene therapy to kill the target cell independent of the PV. Alternatively, small molecules and antisense oligonucleotides can target transcription and translation enhancers to increase protein expression from a remaining wild-type allele, or modulate protein degradation, each ultimately resulting in more functional protein. 27 At the RNA level, target mRNAs may be edited using trans-splicing ribozymes40–44 or SMaRT (Switching Mechanism at the 5′ end of RNA Template) technology,43,45,46 a strategy that does not permanently change the genome. mRNA splicing, as well as miRNA function, can also be modulated with antisense oligonucleotides, to control gene expression in a targeted manner.37,47 While mRNA editing and splicing are also highly personalized, they are well tolerated in pediatric patients with Duchenne muscular dystrophy or spinal muscular atrophy. 48 Nonsense variants, which result in negligible full-length protein expression, can be targeted by nonsense suppression therapy, which uses small molecules that target the translational machinery to restore translation of full-length, functional protein.49,50 Finally, proteins can be replaced, and modifier compounds can be utilized to translocate, stabilize, or otherwise help restore defective function. We consider each modality in detail and elaborate on advantages and disadvantages, their status for NF1/SWN, and examples of clinical success in Table 1.
Approaches for gene-targeted therapies for rare disease and their advantages, disadvantages, status for NF1/SWN, and examples of success.
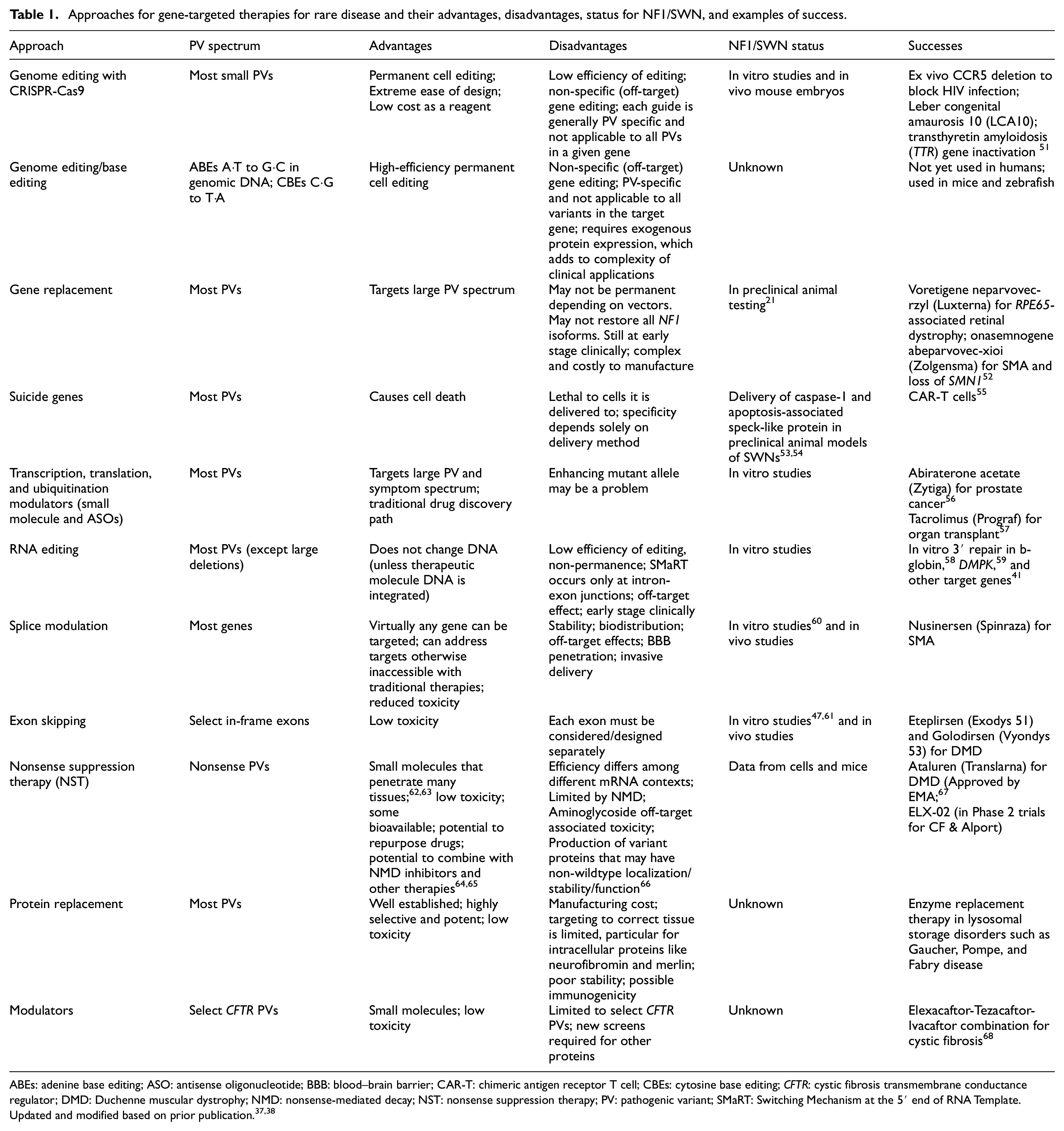
ABEs: adenine base editing; ASO: antisense oligonucleotide; BBB: blood–brain barrier; CAR-T: chimeric antigen receptor T cell; CBEs: cytosine base editing; CFTR: cystic fibrosis transmembrane conductance regulator; DMD: Duchenne muscular dystrophy; NMD: nonsense-mediated decay; NST: nonsense suppression therapy; PV: pathogenic variant; SMaRT: Switching Mechanism at the 5′ end of RNA Template.
Delivery vector platforms
Intracellular delivery of gene-targeted therapy remains a major hurdle in the clinical translation of these treatments, and the safety and efficacy of therapies are largely dependent on the vehicle used to deliver the genetic material or cargo to the target cells. Outside of traditional small-molecule compounds (which travel across the cell membrane due to small size or lipophilicity), other delivery strategies include both viral and non-viral methods. Common viral vectors include adeno-associated virus, adenovirus, lentivirus/retrovirus, and herpes simplex virus 1. 14 Adeno-associated viruses are the most used vectors because they can infect multiple vertebrate species, including humans and non-human primates, can be engineered to increase tissue tropism, and are naturally replication deficient, a distinctive feature that is highly advantageous for their use as delivery vectors. 14 Non-viral methods include cell-penetrating peptides, nanoparticles, and bacteria. We consider each method in detail and elaborate on advantages and disadvantages of each, their status for NF1/SWN, and examples of success in Table 2.
Delivery methods for therapeutics, their applications, advantages, disadvantages, and success.
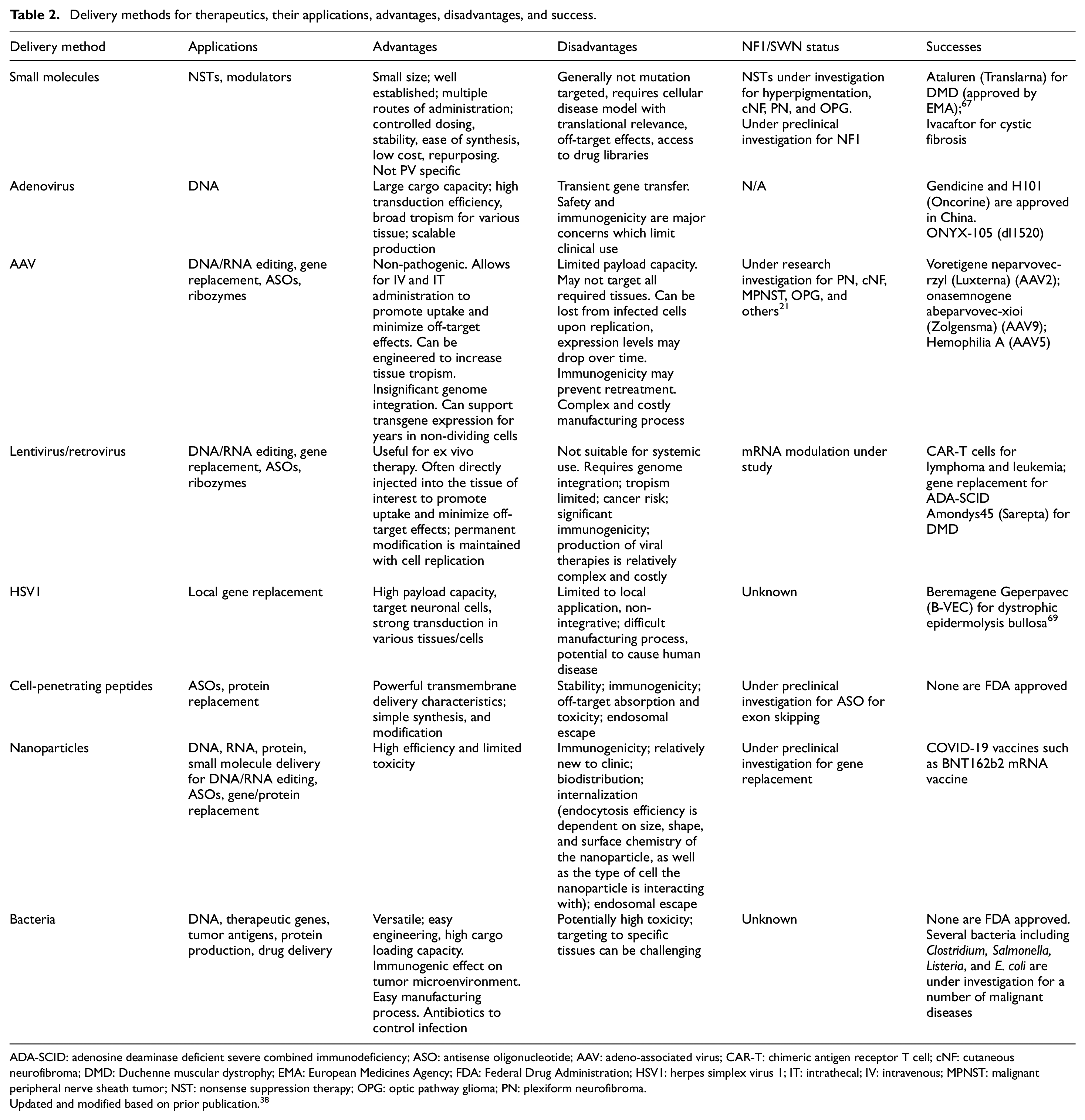
ADA-SCID: adenosine deaminase deficient severe combined immunodeficiency; ASO: antisense oligonucleotide; AAV: adeno-associated virus; CAR-T: chimeric antigen receptor T cell; cNF: cutaneous neurofibroma; DMD: Duchenne muscular dystrophy; EMA: European Medicines Agency; FDA: Federal Drug Administration; HSV1: herpes simplex virus 1; IT: intrathecal; IV: intravenous; MPNST: malignant peripheral nerve sheath tumor; NST: nonsense suppression therapy; OPG: optic pathway glioma; PN: plexiform neurofibroma.
Updated and modified based on prior publication. 38
Some primary delivery concerns include transgene capacity, tissue tropism, and transduction efficiency (the rate or level of transfer of genetic material into the cell of interest). Frequently, the cargo size is a determining factor in choosing the optimal delivery method. For example, the size of the NF1 gene far exceeds the current cargo capacity of adeno-associated virus vectors (∼4.7 kb), thus precluding full-length gene replacement via adeno-associated virus unless using split vector systems or minigene approaches.21,70 Furthermore, since different target tissues may be at the basis of certain phenotypes for both NF1 and SWN, ideally, the therapeutic cargo is targeted to specific cell type(s) to maximize efficacy and minimize off-target accumulation and toxicity. Thus, the tropism or targetability of a respective vector may limit its clinical use to a narrow spectrum of symptoms. Finally, suboptimal transduction efficacy of the target cells may result in outgrowth of untransduced tumor cells.
Preclinical testing in NF1 and SWN models
Ideally, gene-targeted therapy approaches should be tested in animal models that are based on human PVs and that recapitulate tumor natural history as far as growth rate, location, and therapeutic responses (model validation). However, due to the diverse spectrum of NF1 and NF2 PVs in humans, limited understanding of genotype–phenotype correlation, and the type/mechanism of experimentally induced inactivating germline PVs, animals will only partially recapitulate human disease and exhibit a limited spectrum of manifestations (Table 3). Thus, a broad range of models may be needed before advancing to human trials.
Cre-drivers and variants for mammalian NF1 and SWN models.
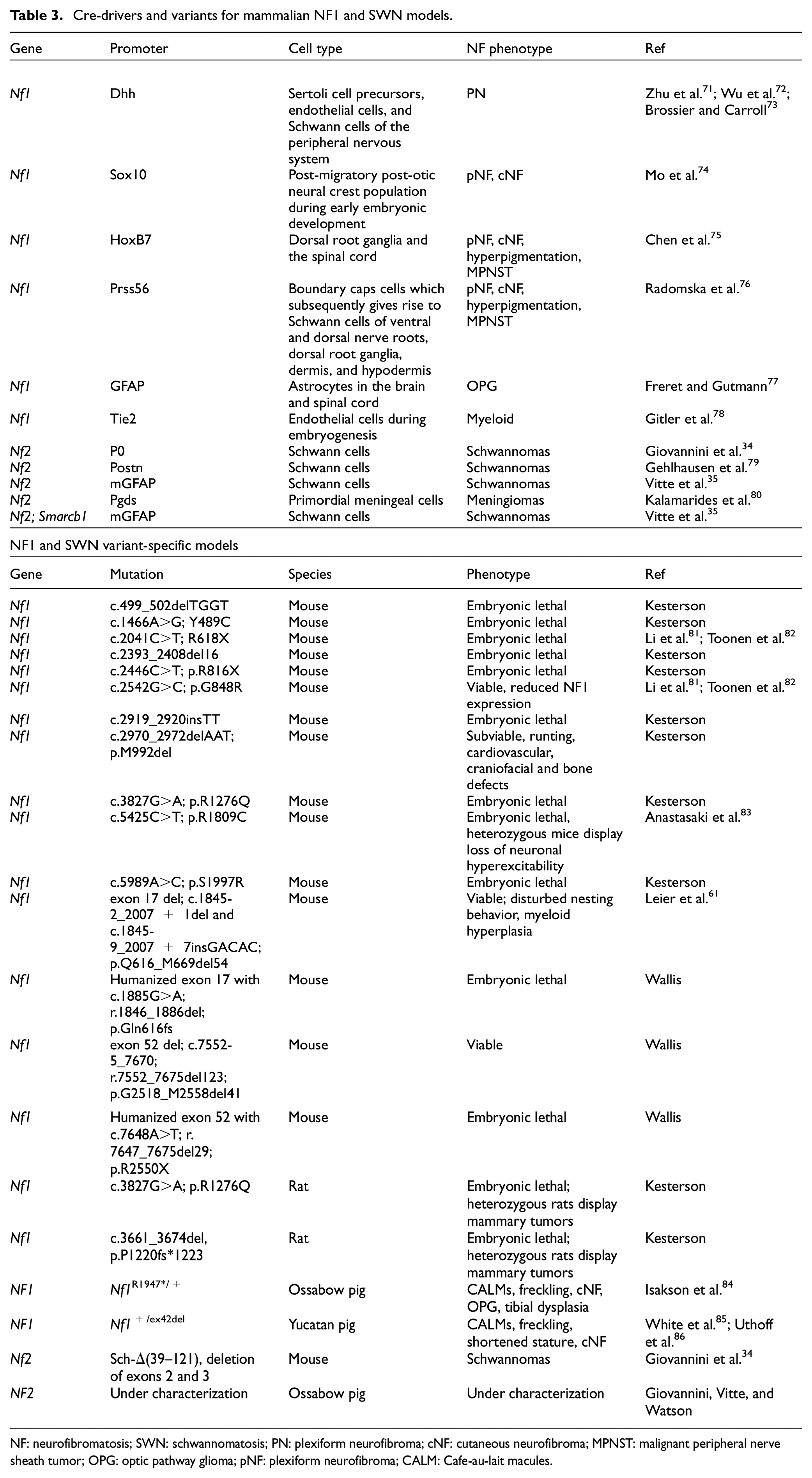
NF: neurofibromatosis; SWN: schwannomatosis; PN: plexiform neurofibroma; cNF: cutaneous neurofibroma; MPNST: malignant peripheral nerve sheath tumor; OPG: optic pathway glioma; pNF: plexiform neurofibroma; CALM: Cafe-au-lait macules.
Murine models have been used in several gene therapy studies as the initial preclinical evaluation platform, including those for cancer, hematological, and neurological conditions. 87 Several mouse models have been designed to mimic loss of heterozygosity at the Nf1 locus (i.e. the second genetic step that results in tumor formation). In general, studies have utilized knockout mice in which gene function is inactivated. However, mice require elaborate genetic manipulations to produce a clinical phenotype by deletion of Nf1 using “conditional” knockouts through cell type and tissue-specific Cre-drivers to produce specific disease features (Table 3). Widely used plexiform neurofibroma models inactivate the Nf1 gene in Schwann cell precursors using Dhh-Cre to produce neurofibromas along the spinal cord;71–73 this model has provided key preclinical data for the development of selumetinib as a therapeutic for plexiform neurofibromas. 88 Cell-specific deletion of Nf1 can also produce cutaneous neurofibromas and plexiform neurofibromas when deleting Nf1 in Sox10- or HoxB7-expressing cells75,74 or in Prss56-expressing cells;76,89 similar strategies can be used to model gliomas, 77 myeloid leukemia, and pheochromocytomas, and non-tumor manifestations such as hyperpigmentation, or bone, vascular, or neurocognitive abnormalities. 90 Next-generation mouse models that recreate patient-specific germline PVs are now available to test therapeutics based on the specific type of genetic alteration; yet, to date, no mouse model fully recapitulates the human phenotype (Table 3).
Several genetically engineered mouse models of NF2-SWN and SMARCB1-SWN have been generated that develop schwannomas with histologic and clinical features similar to humans. Since these models recapitulate the slow-growing and indolent nature of schwannoma, their preclinical therapeutic window is quite large, requiring several months of follow-up. Mouse models based on Nf2-null cell grafts provide a faster readout, but their fast-growing nature due to tumor cell transformation (with acquired additional genetic variants) may bias therapeutic response to merlin reintroduction. Thus, the most relevant model should be chosen based on the translational question. For example, toxicity of gene therapy on NF2-haploinsufficient cells (non-tumor cells) should be analyzed in Cre;Nf2ko/flox models and not in Cre;Nf2flox/flox or allo/xenograft models.
The use of large animal models (pig) with physiology and metabolism closer to humans could improve the predictivity of preclinical testing. 87 Porcine models are well situated to assess different delivery approaches and toxicity studies, as was shown in spinal muscular atrophy and Duchenne muscular dystrophy, 87 whereas lower species (mouse) can be used to address biologic/mechanistic questions. In NF1, two fairly well-characterized porcine minipig models exist, manifesting clinical features of cutaneous neurofibromas, neurocognitive symptoms, and optic pathway gliomas;84,85 in NF2-SWN, one novel porcine minipig model is currently under characterization. Important considerations for use of porcine studies includes high cost, the low prevalence of some disease manifestations (which may mimic humans), and the extended time required for pre-clinical studies (compared to rodent studies).
Finally, non-human primates have played an important role in the evaluation of gene therapies because they are genetically closest to humans. 87 Their nervous systems and anatomy are similar to humans, making them a crucial model for evaluating biodistribution and safety of gene therapies targeting NF1 and SWN; however, non-human primates modeling NF1 and SWN currently do not exist. Hence, not only would utilization of these models to evaluate efficacy require precision model development, but also extensive phenotypic characterization including natural history studies. Notably, non-human primate models were not required for approval of selumetinib and may not be essential for NF-related gene therapy efforts.
Efficacy considerations related to gene-targeted therapy for NF1 and SWN
The therapeutic goals of gene-targeted therapy include stabilization, shrinkage, or prevention of tumors, and treatment of non-tumor manifestations. Given the rarity of these genetic conditions, randomized trials are not practical. Thus, appropriate controls are likely to be derived from Natural History Studies in NF1 and NF2 that have prospectively monitored disease manifestations over time. Selection of participants for clinical trials of gene-targeted therapy will require genotyping to confirm and categorize PVs. Application of the UK NF2 Genetic Severity Score or the revised Functional Genetic Severity Score may help define inclusion criteria and response criteria.
NF1 and SWN are simultaneously monogenic disorders and tumor suppressor syndromes. Effective gene-targeted therapies will need to address several key questions related to the germline and somatic genetic events that cause disease manifestations. For NF1−/−or NF2−/− diploinsufficient tissues (e.g. tumors), can (partial) restoration of neurofibromin or merlin levels shrink or eliminate tumors? If so, how much protein is necessary? At what stage of tumor development? Conversely, for dominant negative variants, what degree of RNA/protein inhibition is necessary to meaningfully effect tumors? Given the range of tumor types in NF1/SWN, will gene-targeted therapies need to be tissue-specific? For NF1+/−or NF2+/− haploinsufficient tissues (e.g. normal Schwann or arachnoid cap cells that can give rise to neurofibromas, schwannomas, and meningiomas after a subsequent somatic inactivating event), can (partial) restoration of neurofibromin or merlin in heterozygous tissues prevent or delay tumor formation? For haploinsufficient tissues associated with non-tumoral disease manifestations (e.g. learning/social deficits, pain, and bone density in NF1; and juvenile cataracts in NF2-SWN), can re-expression of neurofibromin and merlin reverse disease manifestations?
For NF1, targets should initially focus on tumors with the highest morbidity and mortality, such as plexiform neurofibromas, MPNSTs, high-grade glioma, and optic pathway glioma.6–8 Cutaneous neurofibromas, which affect nearly all patients and cause significant disfigurement, may be another attractive target because, unlike plexiform neurofibromas, these tumors are not congenital and typically arise in adolescents and adults rather than young children. These lesions are accessible on the skin, allowing for easy tissue access and monitoring, and may provide opportunities to explore non-systemic administration, as demonstrated by repeated topical application of herpes simplex virus 1 for dystrophic epidermolysis bullosa.69,91 Schwann cells harboring biallelic inactivation of NF1 are considered the tumorigenic cells of neurofibromas and may be targeted with increased expression of wild-type NF1 or transgenes that specifically kill NF1-deficient cells. 17 Alternatively, targeting the haploinsufficient tumor microenvironment may help delay or reverse tumor growth. 27 NF1 gene replacement, nonsense suppression therapy, and modulation of transcription and translation by antisense oligonucleotides are approaches that are well positioned for human studies in the next decade. Genome editing and protein replacement remain attractive options but will require more development before launching clinical trials. NF1 malignancies present additional challenges for gene-targeted therapy. Functional restoration of NF1 alone may not be curative for MPNSTs that harbor additional somatic variants in SUZ12, CDKN2A/B, and TP53, 92 or for high-grade gliomas with somatic variants in TP53 and PTEN. 93
Due to the significant mortality in NF2-SWN, preliminary efforts should focus on this subtype of SWN. 5 As NF2-SWN manifests genotype–phenotype correlations, with constitutional truncating PVs in exons 2 through 13 being most severe, clinical development may focus on targeting these variants, for example via nonsense suppression or gene replacement. 94 Given that biallelic loss of NF2 is common to tumors in NF2-related SWN, LZTR1- and SMARCB1-related SWN, it seems likely that therapies developed in NF2-SWN could also be applicable to other subtypes of SWN. 95 As with NF1, preliminary targets in NF2-SWN are likely to focus on tumors with the greatest morbidity and mortality: intracranial meningioma and vestibular schwannomas. 5 Non-vestibular schwannomas, particularly when considering highly morbid spinal and cranial nerve lesions, are another target of interest. Tumors of the central nervous system may be amenable to localized delivery approaches (intracerebral or intrathecal), and the feasibility of recurrent localized virus administration has recently been demonstrated using oncolytic herpes simplex virus G47Δ, with good safety profile and survival benefit. 96 As with NF1, NF2 gene replacement, nonsense suppression therapy, and modulation of transcription and translation by antisense oligonucleotides are attractive approaches for clinical trials, whereas genome editing is likely to require significant advances prior to human studies.
While symptomatic tumors remain the primary focus for NF1/SWN, a wide range of non-tumor manifestations also significantly impact quality of life and could be explored via gene-targeted therapies. Neurocognitive features of NF1, including learning, attention, and social deficits, affect most individuals to some degree, leading to challenges with academic achievement and social relationships. Brain autopsies and mouse studies demonstrate that neurofibromin loss can alter embryonal migration and differentiation of neuroglial progenitor cells,97–99 and causes pervasive myelin dysfunction, 100 which may limit the efficacy of gene therapies targeting neurocognitive manifestations. On the other hand, downregulation of RAS/ERK signaling in NF1+/− mice rescues attention, working memory, and learning deficits, so neurofibromin upregulation at later stages may still be beneficial.101,102 This is supported by clinical observations indicating that MEK inhibitor treatment may positively impact neurocognitive function in NF1 patients aged from 5 to 27 years. 103 However, given the pediatric onset, variability of neurocognitive manifestations, and possibility for non-NF1-related neurocognitive causes, recruitment may delay human trials. Localized and generalized pain occur in NF1 and SWN, detrimentally impacting quality of life, with variable benefit from medical and surgical interventions. 104 As selumetinib can reduce tumor pain in NF1, it is possible that tumor-directed gene-targeted therapies may show similar benefit. 6
Safety considerations for early-phase clinical trials of gene-targeted therapies
The promise of gene-targeted therapy has been tempered by the historical reality of safety concerns.105–108 Early-phase clinical trials for NF1/SWN must address safety concerns related to specific transgenes and the method of delivery to cells. For gene replacement therapy, it is possible that overexpression of NF1 or NF2 can have negative consequences given their function as tumor suppressors. The comparative effects of enhanced protein expression on NF1/NF2-sufficient, NF1/NF2-haploinsufficient, and NF1/NF2-diploinsufficient cells are unknown, particularly over varying time frames. Other transgenes raise concerns regarding the specificity of on-target effects for tumor cells and off-target effects for non-tumor cells.
The potential short- and long-term consequences of overexpression of neurofibromin or merlin are unknown for both NF1+/−or NF2+/− haploinsufficient tissues (e.g. tissues without a disease phenotype in NF1 or NF2-SWN) and for NF1+/+or NF2+/+ sufficient tissues that occur in persons with mosaic NF1 or SWN. Similarly, the consequences of overexpression of merlin (to shrink or prevent tumors) in non-NF2-SWN patients that have normal levels of merlin throughout their body are unknown. Understanding how overexpression affects these tissues will require the use of clinical trials, and preclinical models can be used to gain valuable insight prior to exposing patients to gene-targeted therapies.
Additional safety concerns for gene replacement are related to viral vectors that integrate into the host genome such as lentiviral vectors. These vectors can activate or inactive nearby genes, giving rise to neoplastic clones. In addition, they can persist in tissues for years after administration, and their effects may be permanent. This property is especially significant for children with NF1/SWN who are severely affected or who are likely to develop severe disease later in life. These children have the most to benefit from gene-targeted therapy but are at highest risk for long-term complications given their expected lifespan. The FDA requires long-term monitoring plans to understand the true safety profile of these products.
In addition, most viral vectors have the potential to elicit an immune response when administered repeatedly. In some situations, this immune response may be desirable (e.g. to stimulate an immune response in a tumor). In other situations, it could limit the efficacy, generate adverse events (i.e. autoimmune disease), or limit the ability to deliver subsequent treatments using the same vector. Given the lifelong risk for developing tumors in NF1 and SWN, retreatment with gene-targeted therapy may be desirable. The use of bacterial or viral vectors also raises the possibility that these can shed in bodily fluids, thereby placing others at infection risk.
Finally, the choice of delivery route is a critical issue and is dependent on the target symptom, vector, location, and indication (prevention vs symptomatic). Intravenous administration may be appropriate for non-tumor manifestations, diffuse disease (e.g. multiple tumors or metastatic MPNST), or when considering symptom prevention. However, intravenous administration necessitates large vector doses to achieve target tissue transduction. High doses may increase toxicity and strain manufacturing capacity, thus slowing clinical translation. 109 Direct administration to the central nervous system via intrathecal injections is more efficient, achieving effective transduction throughout the brain and spinal cord at relatively low vector doses. 110 This approach may be suitable for neurocognitive symptoms and tumors of the brain, spinal cord, and nerve roots. Finally, localized or topical gene transfer may be suitable for solitary and accessible tumors. This approach reduces the risk for systemic toxicity and supports repeated administration of viral vectors owing to reduced immunogenicity.
Conclusion
Gene-targeted therapies hold great promise for NF1/SWN. A key preclinical barrier to success involves defining approaches that can sufficiently target tissues of interest and restore functional protein in correct quantity and at the right time; treatment approaches might differ based on genotype, target symptom, and indication. A key clinical barrier involves identifying a cohort of patients with the appropriate genotype that are at sufficient risk to receive proportionate benefit, while avoiding exposure to individuals that are unlikely going to benefit. As preclinical studies of gene-targeted therapy progress, the wider NF1/SWN community needs to continue discussions surrounding the types of gene-targeted therapies and the potential risks associated with each in order to prepare for early-stage clinical trials.
Footnotes
Acknowledgements
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: V.S. serves as a scientific advisor for the Gilbert Family Foundation Gene Therapy Initiative. H.S. is Founder/CEO of Infixion Bioscience, an early-stage drug discovery company targeting NF1. R.K. serves as a Scientific Advisor for Infixion Bioscience Inc. (3210 Merryfield Row San Diego, CA 92121). Y.K. serves as scientific officer at the Gilbert Family Foundation and as a member scientific advisory panel of NTAP’s Gene Therapy program. S.R.P. is co-founder of NFlection Therapeutics and NF2 Therapeutics and consults for AstraZeneca, SonalaSense, and Akouos. B.K. is a consultant for GenomeMedical, Recursion, Healx, and Springworks.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: V.S. is supported by NTAP’s Gene Therapy Initiative, the National Cancer Institute (NCI) (5K08CA230179 and 1U01CA247576), and the Sontag Distinguished Scientist Award. D.B., J.A.W., A.L., and D.W. are supported by the Gilbert Family Foundation Gene Therapy Initiative. J.A.W. is funded by the Children’s Tumor Foundation’s Gene Therapy Initiative. M.G. and J.V. are supported by NF2 Therapeutics. M.G. receives support from the Department of Defense CDMRP NFRP (W81XWH2110448). J.V. is funded by the Drug Discovery Initiative from the Children’s Tumor Foundation (2020-05-004). B.K. is funded by the NIH and DoD. D.W. and R.K. are supported by the NCI (R01CA26593).