
Abstract
Background:
Informed consent documents for clinical studies should disclose all reasonably foreseeable risks and benefits. Little guidance exists on how to navigate the complexities of risk–benefit communication, especially in early clinical research. Practice-oriented development of such guidance should be informed by evidence on what and how details of risks and benefits are currently communicated.
Method:
We surveyed the responsible parties of phase I/II trials registered in ClinicalTrials.gov that started 2007 or later and completed between 2012 and 2016 to sample informed consent documents from a broad spectrum of early phase clinical trials. Based on an assessment matrix, we qualitatively and quantitatively assessed the informed consent documents for details of risk–benefit communication.
Results:
The risk–benefit communication in the 172 informed consent documents differed substantially in several regards. The outcome, extent, and likelihood of health risks, for example, were described in 83%, 32%, and 63% of the informed consent documents. Only 45% of informed consent documents specified the outcome of mentioned health benefits, and the extent and likelihood of health benefits were never specified. From those informed consent documents reporting risk likelihoods, only 57% added frequency numbers to words such as “common” or “rare,” and even in these cases, we found strong variations for presented frequency ranges. Substantial heterogeneity also exists for how informed consent documents communicate other risk and benefit types and related safeguards.
Conclusion:
Our study points to several shortcomings and heterogeneities in how informed consent documents communicate risks and benefits to potential research participants. Health risks, for example, should be specified with frequency numbers, and health benefits should be specified at least by mentioning their outcomes. Further demand for research and policy development is needed to harmonize risk–benefit communication and to clarify ways to specify the likelihood of health benefits.
Keywords
Background
Informed consent (IC) is an important legal and ethical requirement in research involving human subjects.1,2 Disclosure of the risks and benefits of participating in clinical research is part of all guidance for IC procedures.1,3–6 The US Food and Drug Administration (FDA), 7 for example, requires a mandatory “description of any reasonably foreseeable risks” and “any benefits to the subject or to others which may reasonably be expected from the research ….” The concepts of risk and benefit are complex, and thus, the communication of risks and benefits can involve several elements. The cited guidance does not reflect this complexity and gives no further orientation of, for example, what “any reasonably foreseeable risk” means and what criteria can be used to identify and communicate such risk.
Several empirical studies have assessed the readability and understanding of informed consent documents (ICDs), including risk–benefit information,8–10 or discussed where and why risk–benefit information in ICDs should be reduced or increased.11,12 These studies, however, did not provide a comprehensive assessment of all details for risk–benefit communication such as reported outcomes, extents, likelihoods, and rationales.
While there is no specific matrix for describing all potentially relevant details of risk–benefit information, the International Patient Decision Aid Standards (IPDAS) framework takes a substantial step in this direction, as it outlines essential information units needed to make high-quality decisions. Brehaut et al.13,14 adapted the IPDAS criteria for application on ICDs. The adapted IPDAS framework includes several items relevant to risk–benefit communication. It asks for an overall rating on whether risks (or disadvantages) and benefits (or advantages) are described. Furthermore, it explicitly asks whether the ICD provides information about (1) the probabilities of each benefit and risk, (2) the levels of uncertainty around these probabilities, (3) the reversibility of side effects of participation, and (4) the quality of research evidence used. 15 Brehaut et al. 14 adapted the IPDAS framework to analyze risk–benefit communication in ICDs. Content analyses of ICDs using the IPDAS framework reveal important insights into how often certain details of risk–benefit information are described (probabilities, uncertainty, and so on), but the IPDAS framework does not include other details such as financial risks and benefits, epistemic benefits, or confidentiality risks. The IPDAS framework also does not evaluate what safeguards that aim to reduce/mitigate risks and to optimize/sustain benefits are reported. The Brehaut et al.’s study explicitly excluded discipline-specific risk–benefit information such as research-related injury compensation.
Our study aimed to qualitatively and quantitatively assess what details regarding all different aspects of risk–benefit information are currently communicated in ICDs for phase I/II trials. We focused on phase I/II because this is where uncertainty for harms and benefits is greatest and we were particularly interested to see whether and how ICDs communicate this uncertainty. This study further builds part of a broader project (see section “funding”) that aims to increase quality of ICD development and ethics review for early phase clinical trials.
Methods
Sampling of ICDs
Based on the ClinicalTrials.gov registry, we sampled “phase I/II” trials using the following parameters: (1) “completed,” (2) “interventional” study type, (3) “Anglosphere” countries, (4) start date of 2007 or later, and (5) completed between 2012 and 2016. We focused on completed studies to increase response rates. For the same reason, we excluded all trials lead by private sponsors as we experienced very low response rates in this subgroup in other studies. We aimed to include about 150 ICDs and expected a lower response rate (around 10%–15%) than the Brehaut et al.’s study (expected 20% and yielded 32%) 14 because we are not affiliated with a North-American university what might make responsible persons from North-American universities less likely to participate in our study. We chose the start date of 2007 or later in combination with the above-mentioned completion dates because for this time range we identified 1126 phase I/II trials that met our inclusion criteria. We contacted the “responsible person” for all included trials by e-mail, briefly presented our study design and objectives, and asked the person to send us the trials’ ICD. Our primary request was followed by a reminder after 1, 3, and 7 weeks, based on the tailored design method. 16 To assess the demographic data of all included trials, we downloaded the aggregate analysis of ClincalTrials.gov (AACT) data set in January 2019. To compare demographic data of all included trials with excluded and non-responding trials, we employed the AACT data set downloaded June 2020.
Data extraction and analysis of risk–benefit information
For the purpose of data extraction, we developed an initial assessment matrix for “potentially relevant elements and specifications of risk–benefit information for early clinical research.” The matrix included various elements of risks and benefits reflecting (a) to whom the risks and benefits apply (trial participants or third parties) and (b) the risk and benefit types (e.g. health, reproductive, and financial). The matrix further included specifications that, in principle, apply to all elements of risks and benefits (e.g. likelihood of mentioned risks/benefits and safeguards for reducing risks). The matrix development was informed by established guidelines and scientific literature on risk–benefit assessment and medical decision making.6,14,17 To validate and, if needed, to refine the initial assessment matrix, a random subgroup of 20 ICDs for further elements and specifications of risk–benefit information was screened by three researchers (H.K., S.B., and C.S.) independently.
All ICDs were then read in full and analyzed independently by means of qualitative thematic text analysis 18 by at least one of three researchers (H.K., S.B., and C.S.). To ensure consistent assessment by all researchers, ICD numbers, 1–20, 61–65, 121–125, and 178–181, were independently assessed by all three researchers, compared, and discussed until consensus was reached during the course of extraction and analysis.
After extracting all relevant text passages according to the assessment matrix, we employed descriptive statistics to assess how often ICDs reported or did not report the details of risk–benefit information.
Results
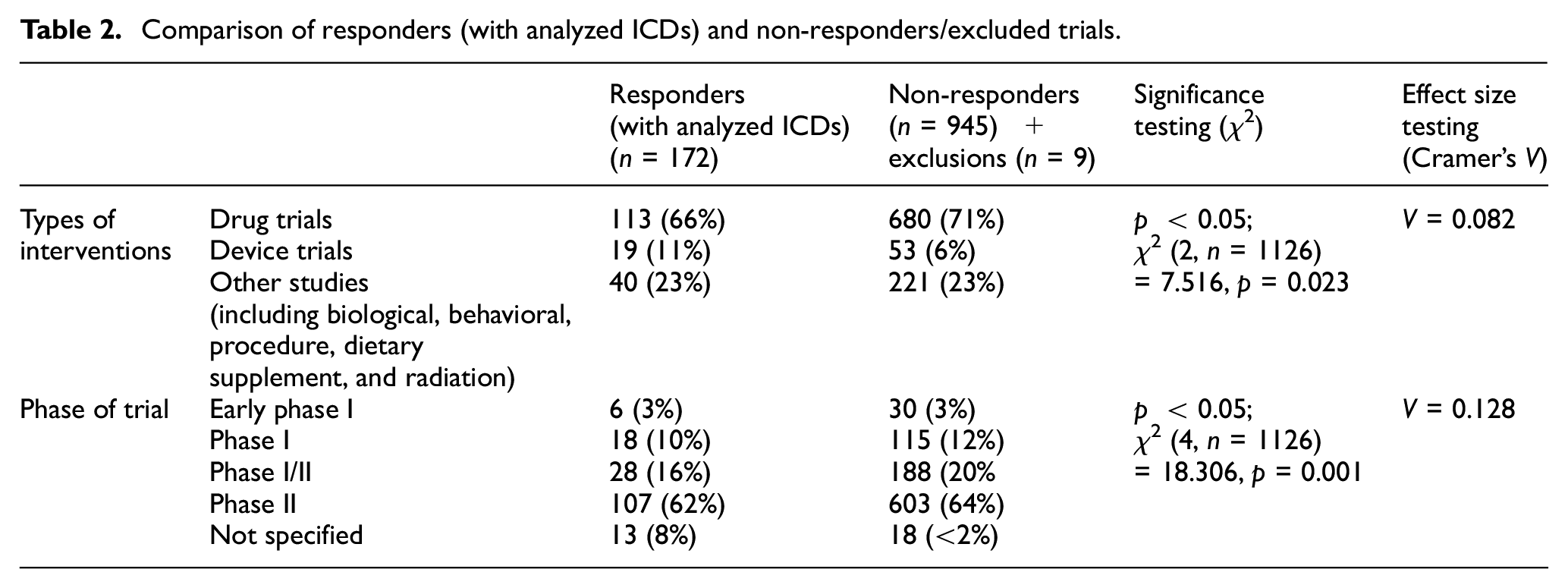
We identified 1126 phase I/II trials in the ClinicalTrials.gov registry that met our inclusion criteria. After contacting the responsible parties of these trials (as indicated in the registry), we received 181 ICDs (response rate = 16%). Some researchers declined to participate in our study for various reasons ranging from legal or ethical concerns to practical obstacles (e.g. no access to the ICD). Nine documents were excluded (see the flowchart in Figure 1). The remaining 172 ICDs (38% phase I/II and 62% phase II) investigated interventions (66% drugs, 11% devices, and 23% others) on 10,861 enrolled persons in total for a wide range of health conditions, including cancer (20%), mental disorders (20%), and cardiovascular diseases (11%) (see Table 1). Most trials were located in the USA (89%) or Canada (6%). Further characteristics of the 172 trials are listed in Table 1. Differences between the 172 responder and the 952 non-responder/exclusions were significant (p < 0.05) but of low-effect sizes for the types of interventions (χ2 = 0.023; Cramers’V = 0.082) and trial phase (χ2 = 0.001; Cramer’s V = 0.128; see Table 2).
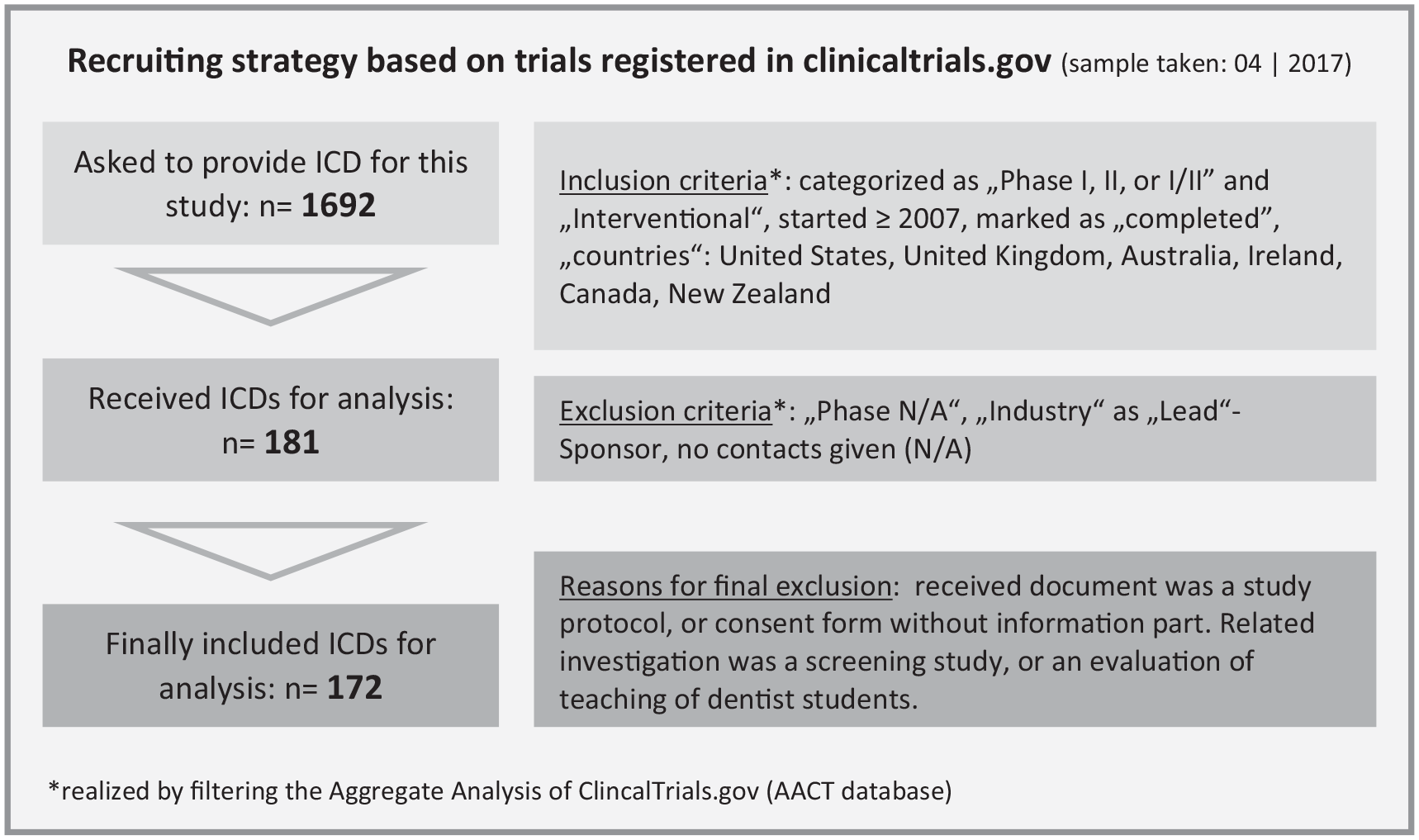
Flowchart of obtaining the data sample (informed consent documents from clinical trials).
Characteristics of trials that provided the analyzed informed consent documents.
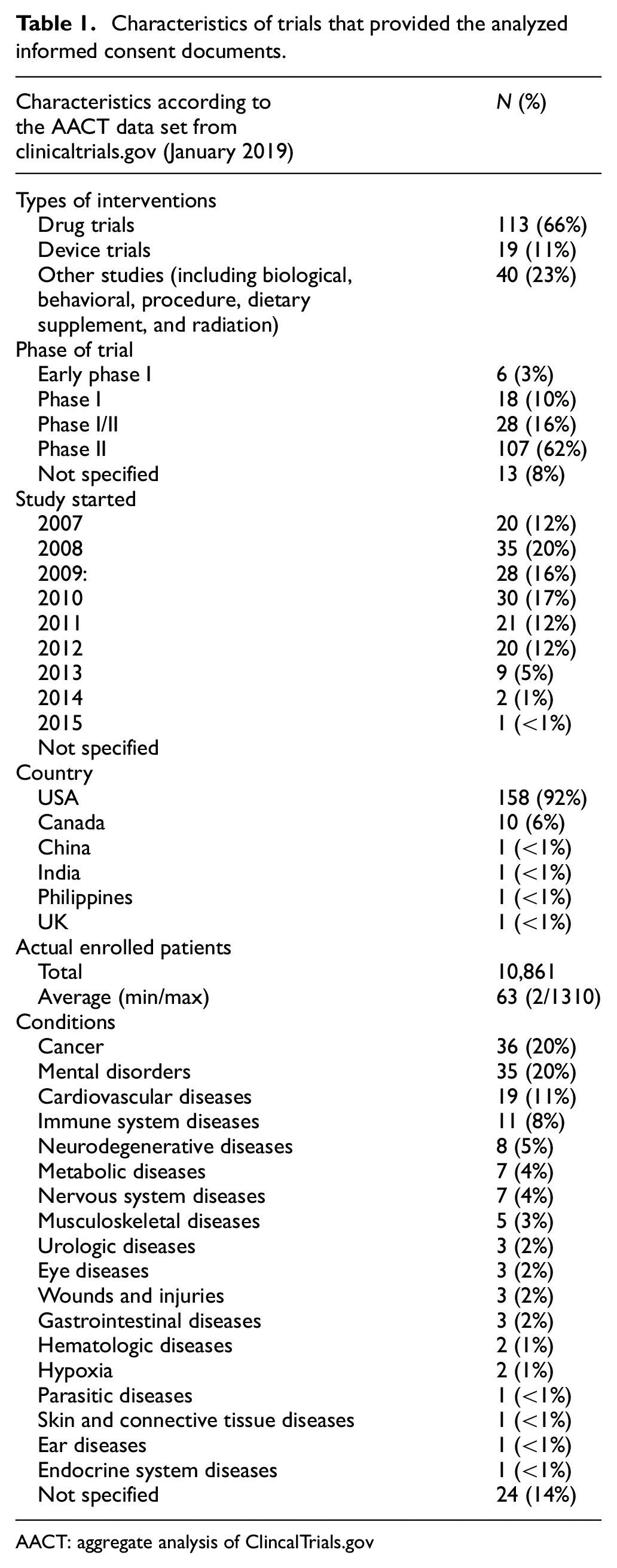
AACT: aggregate analysis of ClincalTrials.gov
Comparison of responders (with analyzed ICDs) and non-responders/excluded trials.
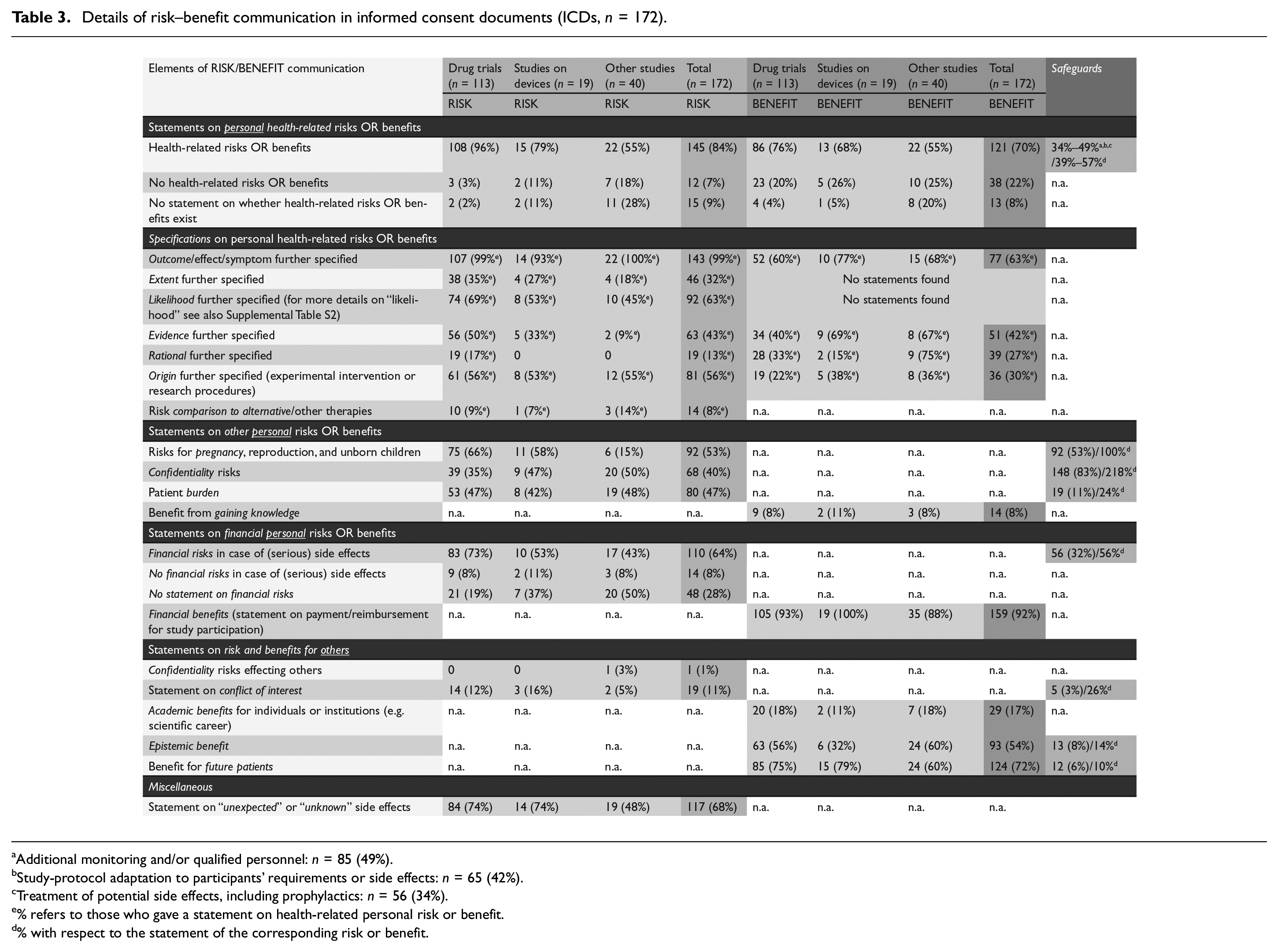
In the following, we describe what details of risk–benefit communication were provided and how often (see Table 3). Supplemental Table S1 presents anonymized text examples for all types of details.
Details of risk–benefit communication in informed consent documents (ICDs, n = 172).
Additional monitoring and/or qualified personnel: n = 85 (49%).
Study-protocol adaptation to participants’ requirements or side effects: n = 65 (42%).
Treatment of potential side effects, including prophylactics: n = 56 (34%).
% refers to those who gave a statement on health-related personal risk or benefit.
% with respect to the statement of the corresponding risk or benefit.
Information on personal health risks or benefits
Of all 172 ICDs, most mentioned that some health risks (n = 145, 84%) and benefits (n = 121, 70%) may exist for those participating in the respective trial. Among the subgroup of drug trials, these numbers were higher (96%, n = 108 and 76%, n = 86). Table 3 presents the results for all subgroups. Of all 172 ICDs, 12% and 38 (7% and 22%, respectively) mentioned explicitly that no health risks and benefits existed, respectively. See Table S1 for respective text examples. Of all 172 ICDs, 15% and 13 (9% and 8%, respectively) included no statements on whether health-related risks and benefits existed.
Health risks were mainly communicated through a few typical wordings, such as “Risk xyz may occur/could happen,”“Reported side effects are xyz,” or “There is a risk that xyz.” The 113 drug trials mentioning health benefits for the participants most often employed wordings such as “you may or may not benefit from participating in the trial” (n = 62, 69%), other wordings included “we hope that you will benefit,”“we cannot guarantee that you will benefit,”“you should not expect any benefit,” or “it is not possible to predict whether you will benefit.”
The majority of ICDs further specified the outcome of the respective risks (n = 143, 83%), while only 77 ICDs (45%) were equally specific for the outcome of the respective benefit (EXPLs 1; Table S1). Further rationales for why certain risks and benefits are expected were mentioned in 13% and 27% of all ICDs, respectively (EXPLs 5; Table S1). Supporting evidence for the expectation of risks and benefits was mentioned in 43% and 42% of all ICDs, respectively (EXPLs 4; Table S1).
Of all 172 ICDs, 32% (n = 46) provided further information on the extent of risks and 63% (n = 108) specified the likelihood of risks (EXPLs 2 and 3; Table S1). No ICD specified the extent or likelihood of the mentioned benefits. Of the 108 ICDs that mentioned risk likelihoods, 43% communicated likelihoods only by qualitative wordings such as “common” or “more likely,” while the other 57% combined qualitative wordings with relative numbers (see Supplemental Table S2 for more detailed information).
Most ICDs (55%) employing qualitative communication of likelihoods used the following gradation: (a) most common/likely, (b) common/likely, (c) less common/likely, and (d) rare. We found strong variations when ICDs specified qualitative risk communication using numbers. For events that were expected to occur in 15%–20% of the cases, for example, we identified three different wordings used: “most common” (ICD 9), “common” (e.g. ICD 30), and “less common” (e.g. ICD 66). Table 4 presents all identified ranges. In addition to the four gradations mentioned above, some ICDs used other terms, such as “very likely (25%),”“more common (5% or more),”“frequent (10%–50%),” and “uncommon (1%–5%).”
Range (relative numbers) used for the gradations of frequencies/likelihood of risk.
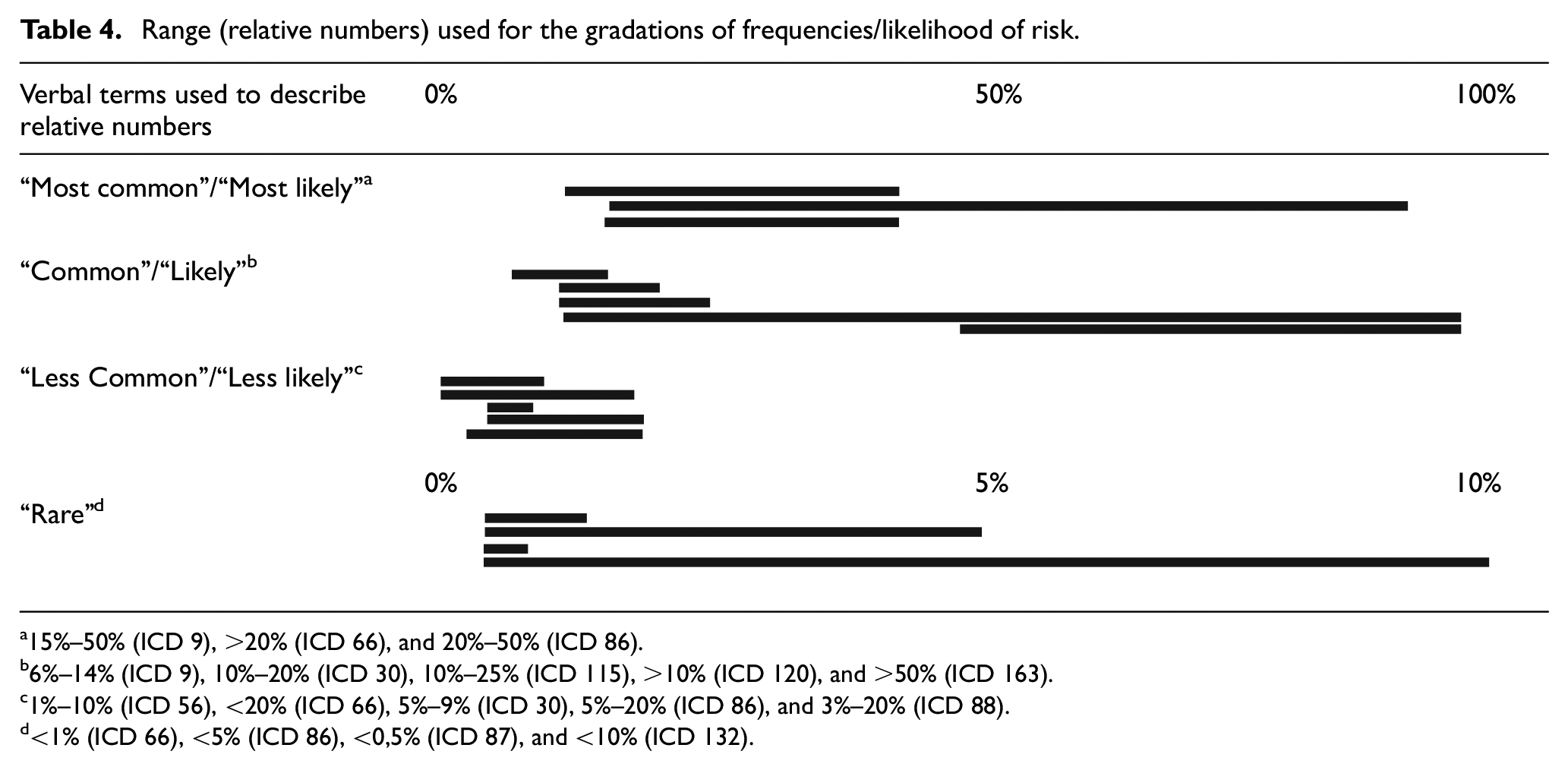
15%–50% (ICD 9), >20% (ICD 66), and 20%–50% (ICD 86).
6%–14% (ICD 9), 10%–20% (ICD 30), 10%–25% (ICD 115), >10% (ICD 120), and >50% (ICD 163).
1%–10% (ICD 56), <20% (ICD 66), 5%–9% (ICD 30), 5%–20% (ICD 86), and 3%–20% (ICD 88).
<1% (ICD 66), <5% (ICD 86), <0,5% (ICD 87), and <10% (ICD 132).
We also assessed the origin of the risks and benefits. The origin of risks and benefits can be the investigational (therapeutic) treatment or additional (non-therapeutic) research procedures. The origin of risks and benefits was mentioned in 56% and 30% of all ICDs, respectively (EXPLs 6; Table S1). The risk comparison to alternatives was mentioned by 8% of all ICDs (EXPLs 7; Table S1).
Table 3 presents more detailed findings for three types of study interventions (drugs, devices, and others).
Information on other personal risks or benefits
Of all ICDs, 40% (n = 68) explicitly mentioned confidentiality risks and 64% (n = 110) mentioned some financial risks (e.g. costs for treating study-related injuries or disadvantages for insurance (e.g. life insurance) or employment). Another 8% mentioned that no financial risk for health care exists. The remaining 48 (28%) did not mention whether financial risk exist.
Information on risks or benefits for third parties
With regard to benefits for third parties, 72% mentioned potential benefits for future patients and 54% mentioned epistemic benefit (for science and society) (EXPLs 11 and 12; Table S1). Other elements were covered by only a few ICDs such as statements of conflicts of interest (11%) or academic benefits in the form of perspectives for a scientific career (17%) (EXPLs 9 and 10; Table S1).
Further findings for other details of risk–benefit information are presented in Table 3.
Safeguards
Across all 172 ICDs, we identified three types of safeguards for reducing personal health risks: (a) additional monitoring and/or qualified personnel (mentioned in 49% of all ICDs), (b) study-protocol adaptation to participants’ requirements or side effects (42%), and (c) treatment of and prophylactics to prevent potential side effects (34%) (EXPLs 14 and 15; Table S1). All ICDs disclosing risks for pregnancy, reproduction, and unborn children also mentioned safeguards such as the recommendation of pregnancy testing, the exclusion of pregnant women, or advice to use contraception. In contrast, for all ICDs mentioning conflicts of interest, only 26% mentioned a safeguard (EXPL 16; Table S1).
Typically, as for all above described risk-safeguard pairs, the ICDs mentioned safeguards only after mentioning the respective risk. For confidentiality risks, we identified another pattern. While 40% of all ICDs explicitly mentioned confidentiality risks in combination with an accompanying statement on safeguards, such as data protection, another 43% of all ICDs mentioned data protection or other safeguards without explicitly mentioning the confidentiality risk (EXPLs 18 and 19; Table S1).
With regard to safeguards ensuring benefits, we identified four ICDs (2%) that mentioned their registry entry at ClinicalTrials.gov, seven ICDs (5%) that specified their plans to translate the results of the planned study into future research. We found no statements regarding planned open data, that is, whether secondary use of patient-level data assessed within the trials will be possible for other research questions studied by other research groups. Table 3 presents more findings on statements dealing with safeguards.
Discussion
To facilitate voluntary participation in clinical research, ICDs should adequately inform potential trial participants about the anticipated benefits and risks of the study. While most experts and laypersons will probably agree that a simple statement such as “Participating in this clinical trial may come with risks and benefits” is insufficient and thus inadequate, much less agreement exists on what more detailed information in ICDs makes risk–benefit communication adequate. In this study, we assessed what details about study-related risks and benefits 172 ICDs for phase I/II trials communicate to potential participants. We structure the discussion of our results into three parts: (1) shortcomings in risk–benefit communication, (2) questionable heterogeneity, and (3) normative-conceptual issues in need of further debate.
Shortcomings in risk–benefit communication
Only 63% of all ICDs communicate the likelihood of reported risks. Of those ICDs reporting risk likelihoods, 43% employed only qualitative risk information despite evidence-based recommendations to present risk likelihoods numerically.19,20 Even when qualitative wording is specified by frequency numbers, we found strong variations for how wordings such as “most common,”“less common,” or “rare” are specified. A broadly established convention for frequency groupings is: very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1000 to <1/100), rare (≥1/10,000 to <1/1000), and very rare (<1/10,000). 21 A shortcoming in benefit communication is that of all ICDs mentioning health benefits (n = 121), only 63% specified the benefit outcome. All ICDs should specify the quality of the outcomes of both risks and benefits. It makes a difference whether a trial aims to reduce tumor growth, improve quality of life, or cure the disease.
Questionable heterogeneity
We found a strong heterogeneity in how the ICDs reported other potentially relevant details in risk–benefit communication. For example, 26% of ICDs provided a rationale for why the health benefits are anticipated. Likewise, 40% of all ICDs went a step further and mentioned supporting evidence (preclinical or clinical) for the anticipated benefit. When and whether at all supporting evidence should build part of risk–benefit communication in ICDs can be discussed controversially. Only few study participants would have context for evaluating such information, but some case analyses demonstrated the potential relevance of providing such information in certain situations.22,23 Further analysis needs to inform the development of guidance on when and how ICDs should communicate supporting evidence. Based on such guidance, further in-depth empirical studies could assess when ICDs report too much or too little information in this regard.
A similar mix of heterogeneity and insufficient guidance exists for communicating the origin of risks. Only half of all ICDs mentioned whether risks originate from the study intervention or from non-therapeutic research procedures. It might make a difference for potential trial participants whether substantial risks originate from potentially beneficial interventions or from invasive non-therapeutic research procedures. More guidance is needed on when and how to communicate the origin of risks. An expert group recommended to structure the consent process for oncology trials in a way that enables potential participants to acknowledge and accept (non-therapeutic) research procedures and that does not distract them by a focus on drug risks. 24
Furthermore, very few ICDs communicated safeguards for ensuring the trials’epistemic benefits (e.g. preregistration and data sharing) or the risks and benefits of alternative interventions (Table 3). Again, explicit guidance on when and how ICDs should communicate these details of risks and benefits would be helpful for those who write and review ICDs.
Normative-conceptual issues in need of further debate and guidance
While best-practice guidelines for risk–benefit communication in health care clearly recommend presenting likelihoods for both risks and benefits, this is not an established standard in ICDs. Of the 121 ICDs that mentioned the potential of health benefits, no ICD (0%) specified the respective likelihoods. The most often employed and very vague wording was “you may or may not benefit.” Other identified wordings such as “we hope that you will benefit” or “we cannot guarantee that you will benefit” might also introduce overoptimistic estimates regarding personal health benefits. King and colleagues25,26 described this situation already two decades ago and commented that this likelihood language might reflect a struggle between two conflicting impulses of principal investigators: to not overpromise and to not take away hope. Next to several expert recommendations also the NIH Guidance on Informed Consent For Gene Transfer Research explicitly states that these simple potential benefit statements “do not provide sufficient meaningful information for potential participants” and that such statements “should be avoided whenever it is possible to provide more specific information about the nature and likelihood of potential benefits.” 27
More explicit and specific communication on benefit likelihoods might be possible, especially in drug research. ICDs could, for example, inform participants about past success/failure rates in trials with the same drug or in trials in the same therapeutic area. More recently, discussion has started around considering “portfolio-level” information on success and failure rates in cases where an already approved drug is tested for other indications. Mattina et al., 28 for example, showed that after two clinically useful applications the cancer drug sorafenib was discovered in the first two efficacy trials, the drug was tested for 26 further indications and in 67 drug combinations, leading to only one additional licensure. 29 Should ICDs written for trials investigating the 26th indication inform participants about the fact that the drug was effective for the first two indications but failed for the 23 indications that followed?
The likelihood of approval (LOA) could serve as another concept to inform participants about success rates, this time within therapeutic areas. The LOA of new drugs entering phase II trials ranges between 11% (in oncology) and 25% (in infectious diseases).30,31 Furthermore, the German health technology assessment (HTA) agency (IQWiG) reports that of all 288 newly approved drugs that they assessed in 2018, less than 30% (oncology = 38%) showed a medium- or high-additional benefit. 32 Would it be more appropriate if ICDs for phase II oncology trials include statements such as “you should not expect to benefit because, over the past five years, only 4% of new oncology drugs tested in trials like this one ultimately demonstrated a medium- or high-additional benefit over standard treatment”? Put differently, why would it be more appropriate to communicate merely that “you may or may not benefit” if we know that past statements reflected a 4% chance of benefit? In a recent paper, Zhang et al. 33 further specified the concept of LOA by measuring how often patients in phase I cancer trials received a treatment that was approved for their indication at the doses received. The authors conclude that estimates on the proportion of patients in phase I trials, who access a drug that will be approved for their conditions, provide a basis for communicating risk and benefit to patients in phase I research.
A third source for generating quantitative information about potential benefit could be the expected effect size that directed the sample size calculation. The expected effect should be at least as large as the minimal clinically relevant effect. 34 The estimation of effect sizes is based on literature searches, pilot studies, or expert judgments. Whether such estimates are of sufficient procedural and outcome quality demands further research. 35
The communication of the expected likelihood or extent of health benefits in clinical trials would be a complete novelty in ICDs. Further research and discussion are needed to identify the strengths and weaknesses of the three above-mentioned sources for patient-oriented risk–benefit information and their communication in ICDs for clinical trials.
Our study has the following limitations. First, our sample did not allow subgroup analysis of, for example, high-risk research (e.g. gene transfer trials) or certain therapeutic areas. We chose a more diverse sample to increase the probability that we capture all currently reported types of details for risk–benefit communication (elements and specifications). Furthermore, our sample included only ICDs of completed trials. We believe that sampling ICDs of ongoing phase I/II trials would result in a too low response rate. Our sample, therefore, included ICDs that were employed for studies starting from 2007 to 2013. Because no new laws or guidelines that focus on risk–benefit communication in ICDs were published over the past decade, we do not see reasons to believe that the patterns described in this article changed substantially. Our sample mainly covers trials conducted in the USA (89%) and Canada (6%). While national regulations are very similar regarding the reporting of personal health-related risks and benefits in ICDs, reporting on financial risks can be different in countries with statutory health insurance. For reasons described in section “Methods,” we excluded trials lead by private sponsors. Future studies should evaluate whether ICDs from industry trials present different patterns for the described shortcomings and heterogeneity.
Conclusion
In summary, we believe that risk–benefit communication in ICDs for phase I/II trials should and can be improved without increasing the lengths of ICDs. Our analysis demonstrated that the communication of risk likelihoods should be improved by following broadly established guidance. Short but explicit statements on the risks of alternative treatment options and standard care would also improve risk communication without dramatically increasing the lengths of ICDs. Further practice-oriented work (e.g. via templates, checklists, and automated text analysis tools) is needed to support researchers and review-bodies in improving the quality of risk–benefit communication in ICDs. An important but hitherto neglected discourse is needed on how to improve the communication of benefit likelihoods in ICDs. We briefly introduced potential candidates such as (dose-specific) LOA, portfolio-level evidence for success rates, and benefit estimates reflected in sample size calculations. However, further normative and conceptual debate is needed to specify and balance the strengths and weaknesses of introducing information on benefit likelihoods in ICDs.
Supplemental Material
sj-docx-1-ctj-10.1177_1740774520971770 – Supplemental material for Details of risk–benefit communication in informed consent documents for phase I/II trials
Supplemental material, sj-docx-1-ctj-10.1177_1740774520971770 for Details of risk–benefit communication in informed consent documents for phase I/II trials by Hannes Kahrass, Sabine Bossert, Christopher Schürmann and Daniel Strech in Clinical Trials
Footnotes
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article:
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the German Federal Ministry of Education and Research (GEENGOV-Governance of research using genome editing technology in humans, No. BMBF 01GP1611A, ![]() ). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the article.
). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.