
Abstract
Objectives
Phytic acid (PYT) also known as inositol hexakisphosphate or inositol polyphosphate has shown a broad range of biological effects including anti-inflammatory, antioxidant, and anticancer effects in several preclinical studies. This study aimed to investigate the effects of PYT in vitro on HCT116 and HT-29 cell lines and to analyse the intricate mechanism of NF-κB-β-catenin signalling pathways.
Material and methods
Both cell lines were treated with PYT, and analysed for cell viability, apoptosis, progression of cell cycle, and DNA fragmentation. Gene and protein expression analysis was performed to assess the molecular mechanism.
Results
PYT suppressed the proliferation of colorectal cancer cell lines in a dose- and time-dependent manner with an estimated IC50 value of 2.96 and 3.35 mm, respectively. PYT caused cell cycle arrest at the G2/M phase in both CRC cell lines and induced mitochondrial intrinsic apoptosis via activation of caspase-9 and caspase-3 cascade. PYT suppressed the expression of pro-inflammatory markers especially COX-2 and iNOS, and IL-lβ, IL-6, and IL-10. Analysing the mechanism behind the effects of PYT showed that it suppressed the levels of NF-κB and β-catenin and inhibited the levels of cyclin Dl and c-Myc (its downstream targets) and COX-2.
Conclusion
The results collectively indicate the potent anti-inflammatory and anti-proliferative effects of PYT in CRC cell lines that were mediated by downregulating the β-catenin and NF-κB signalling pathways. Results advocate that natural supplementation of PYT can be an effective preventive approach in controlling cancer of colorectal region.
Introduction
Cancer cell proliferation and metastatic transformations are principally regulated through several cellular disruptions including altered intracellular signalling networks engaged in the transduction of signals in abnormally or overexpressing ways.1–3 These networking alterations allow to focus on the approach of targeting uncontrolled cancer cell signaling cascade through a coherent chemoprevention mechanism. Aberrant expression of genes in cell growth regulation and chronic inflammation pathways have been critically associated with several multistep molecular events in colorectal carcinogenesis and metastasis. These alterations in particular dysfunctions of the immune system, overexpression of cytokines and chemokine, and production of reactive oxygen species (ROS) collectively work in a crosstalk that affects cancer cell proliferation. 4
Colorectal cancer (CRC) is the second most leading cause of cancer associated mortalities worldwide. Amongst the pathogenic events regulating CRC progression, overexpression of several growth and inflammatory signaling pathways are critically associated. Furthermore, chronic inflammation is recognized as the major risk factor for the progression and growth of human cancers in various organs including gastrointestinal cancers like CRC.5,6 Colorectal tumors mostly arise in sporadic forms and only 5–10% of cases are heritable familial adenomatous polyposis (FAP).4,6 CRC progression is resultant of chronic or severe exposure to chemicals and xenobiotics modulating cellular inflammation signalling which limits the rate of apoptosis. Therapeutic and prevention research focussing on CRC mainly intervenes through regulation of the molecular cross-talks between inflammation and cell proliferation signalling pathways. This specifically involves the network uniting on the redox-sensitive transcription factor NF-κB and growth signalling pathway Wnt/β-catenin. 7 NF-κB has been intrinsically correlated with inflammatory retorts in cells extending toward carcinogenic processes.2,3,8,9 It has been known to mediate inflammation-driven carcinogenic transactivation of cyclooxygenase-2 (Cox-2) as a major molecular target. 10 Cox-2 is associated with the biosynthesis of prostaglandins involved in inflammation and upregulation of several premalignant and malignant transformative processes. 10 Overexpression of Cox-2 has been contributory to tumorigenesis in several types of cancers and its downregulation has been consequently preventive in nature.11–13 The Wnt/β-catenin signaling pathway regulates the transcriptional activity of key developmental genes (cyclin D1 and cMyc) towards cell growth and proliferation. 14 β-Catenin functions as a key mediator of the canonical signalling pathway helping in cell-cell communication through cell adhesion function on the plasma membrane. 15 The cytoplasmic level of β-catenin is controlled via its proteosomal destruction that involves several proteins mainly GSK3 and APC. APC is a tumor-suppressor gene that negatively regulates the function of β-catenin, and thus APC directly affects the progression of both hereditary (FAP) and sporadic forms of CRC. 16 Aberrant activation of β-catenin due to inactivation of its destruction complex or by other means causes transcriptional over-activation of numerous target genes that mainly includes cMyc and cyclin D1.16,17 These two genes are most frequently activated in the case of CRC and further contribute to the growth, invasion, and metastasis of CRCs. 17 Therefore, targeting NF-κB dependent inflammation pathways and Wnt/β-catenin regulated cell growth signalling pathways are key therapeutic regulators in CRCs. Reports suggest that diminishing NF-κB-COX-2-β-catenin crosstalk may prevent tumor growth and that applying natural products may be successfully utilized in interfering with this crosstalk. Several natural products and their lead molecules have been utilized for the modulation of activities of NF-κB and COX-2 in a variety of cancers.18–21 The conventional therapeutic measures against inflammatory carcinogenic process that mainly includes non-steroidal anti-inflammatory drugs (NSAID) act by blocking the prostanoid synthesis from arachidonates through inhibition of the Cox-2 activity.8,10 Yet several adverse complications are associated with their applications. Thus, natural therapeutic and preventive agents with lesser side effects and targeting specific signalling pathways are explored.
Phytic acid (PYT) is a dietary sourced inositol (IP6) or inositol polyphosphate or inositol hexakisphosphate mainly found in fruits and foods like variety of cereals, legumes, seeds, nuts and oil, and soybeans. PYT has been reported to act as a potential inhibitor of colon carcinogenesis.22,23 The intake of dietary fibers containing PYT has shown a decreasing trend in the incidence of CRC which suggests its chemopreventive abilities.24,25 Phytate contributes for decreasing cell proliferation and tends to suppress tumor growth as reported in notable studies. PYT from rice bran showed antiproliferative and antioxidant effects on several cancer cell lines. 26 Phytic acid remarkably repressed the expression of iNOS and NF-κB in activated microglial cells featuring inflammatory responses. 27 IP6 attenuated the levels of IL-6 and IL-8, and NF-κB and IKBα in Caco-2 cells upon stimulation with IL-1β. 28 IP6 controlled the growth of advanced PCA cells and induced apoptosis by inhibiting the expression of constitutively activated NF-κB. 29 A long-chain unsaturated acyclic alcoholic diterpene phytol was shown to have cytotoxic, anti-inflammatory, antinociceptive, antioxidant, anxiolytic, apoptosis and autophagy inducing, immune-modulating, metabolism-modulating, and antimicrobial effects via mechanisms involving modulation of NF-κB and other inflammation and cell proliferation mediators.30,31 One of the plausible mechanisms of PYT is to interplay the regulation of β-catenin and COX-2 and exert a significant part in the inhibition of CRC cell proliferation. 25 This study investigated the effects of PYT on the proliferation of CRC cells and attempted to identify the mechanistic interaction of NF-κB-COX-2-β-catenin signalling crosstalk.
Materials and methods
This study was conducted as a ‘cell culture based preclinical study’ which has no requirement of Ethical Approval.
Cell culture and test drug treatment
Human CRC cell lines HCT116 (ATCC CCL-247; carcinoma, colorectal) and HT-29 (ATCC HTB-38; adenocarcinoma, colorectal) were maintained in RPMI 1640 culture medium (Thermofisher, USA). The media was supplemented with 10% fetal bovine serum (FBS), 2 mm L-glutamine, and penicillin (100 units/ml) and streptomycin (100 μg/mL). The cell culture was incubated in a humidified atmosphere of 5% CO2 in a CO2 incubator at 37°C. Cells were treated with PYT (Sigma-Aldrich, MO, USA) at variable concentrations in vehicle control DMSO at less than 0.1% final concentration. Cells were treated with vehicle control or PYT for 48 h for cell viability assay and for 24 h for further experiments. DMSO was utilized as vehicle control at a concentration less than 0.1% which is considered to be a non-toxic concentration for cell cultures and commonly used in such experiments.
Cell viability and clonogenic assay
HCT116 and HT-29 both cell lines were seeded in 96-well plates at the count of 4000 cells per well for 24 h at 37°C in a CO2 incubator. Cells were treated with indicated concentrations of PYT for 48 h. Cell viability was assayed by using the MTT cell viability assay kit (Thermo Fisher, USA) as per the manufacturer’s instructions. The absorbance of color developed was recorded using a microplate reader at 570 nm. The viable cell percentage was calculated against the percentage of viable cells in vehicle control (100%). For the clonogenic assay, HCT116 and HT-29 cells were treated with PYT at 2.5- and 3.5-mm concentrations for 24 h. Cells were stained with crystal violet for 60 min at 37°C following standard procedure and visualized under a light microscope at 40× magnification. Each experiment was perfumed in triplicates or three experimental repeats.
Flow cytometry for apoptosis and cell cycle analyses
HCT116 and HT-29 both cell lines were treated with PYT at 2.5 and 3.5 mm and vehicle control for 24 h. The apoptosis pattern was analysed using a dual Apoptosis staining kit of Annexin V-FITC (BD Biosciences, USA) according to the manufacturer’s instructions. Flow cytometry of FITC-conjugated annexin V and propidium iodide (PI) and analyzed by the FACSCalibur program. The histogram generated was gated into four standard quadrants designating the population of cells (in percentage) of normal cells (bottom left), early apoptosis cells (bottom right), late apoptosis cells (upper right), and necrotic cells (upper left). A bar graph representing the percentage of cells in normal and apoptosis phases (early and late) was drawn to compare the results in a quantitative manner.
For cell cycle analysis, HCT116 and HT-29 cell lines at 2 × 106 densities were treated with PYT at 2.5 and 3.5 mm and vehicle control in a 6-well plate for 24 h at 37°C. Cells were harvested, stained with trypan blue and counted for analysis. A total of 1 × 106 cells were pelleted by centrifugation and fixed in cold ethanol (70%) for 60 min and placed at 4°C. Cells were finally resuspended in 1 mL Krishan’s buffer supplemented with propidium iodide (50 μg/mL). Incubated the cells for 60 min at 4°C and then washed with cold PBS. Cells were analysed by flow cytometer using the FACSCalibur program, and the percentage of cells in different stages of the cell cycle (G0/G1, S, and G2/M) was estimated by the Modfit LT program.
DNA fragmentation analysis for apoptosis
HCT116 and HT-29 cells were treated with PYT at 2.5 and 3.5 mm and vehicle control for 24 h. Genomic DNA was extracted and agarose gel electrophoresis for DNA fragmentation was performed. The cells were lysed in cell lysis buffer and centrifuged at 12,000×g for 15 min at 4°C. Supernatant was collected and treated with an RNA digestion solution followed by protein digestion reagent. Genomic DNA was isolated from cells and separated onto 1% agarose gel electrophoresis with staining of the DNA with ethidium bromide. The bands of DNA were visualized under a gel documentation scanner.
Mitochondrial membrane potential assay
HCT116 and HT-29 cell lines were treated with PYT at 2.5 and 3.5 mm and vehicle control for 24 h. Mitochondrial membrane potential (MMP or ΔψM) was assayed by using JC10 MMP Assay Kit (Abcam, USA) for microplate as per the manufacturer’s instructions. Cells were stained with JC-10 dye-loading solution (50 μL/wells) and incubated in a CO2 incubator for 30 min at 37°C. Then fluorescence intensities of monomeric forms and J-aggregates were measured at Excitation/Emission wavelengths (490/525 and 490/590 nm). The relative fluorescence units (RLUs) of samples were presented as percentages as compared to vehicle control (100%).
Protein isolation and western blotting
HCT116 and HT-29 both cell lines were treated with PYT at 2.5 and 3.5 mm and vehicle control for 24 h. Total cellular protein was isolated from cells by harvesting them from plates by scrapping. Cells were washed with cold PBS and collected to pellet by centrifugation. Cells pellet were lysed using cell lysis buffer and the homogenate was centrifuged to collect clear supernatant. Protein content in samples was assayed by using the BCA protein assay kit using the standard methodology. An equivalent amount of protein (20 μg) per sample was electrophoresed by SDS-PAGE and blotted onto the PVDF membrane. Membranes were blocked with 5% fat-free dry milk followed by treatment with specific primary antibody. Then the membranes were treated with respective secondary antibodies and proteins were detected by using ECL detection system.
Quantitative real time-PCR (qPCR) from gene expression analysis
HCT116 and HT-29 both cell lines were treated with PYT at 2.5 and 3.5 mm and vehicle control for 24 h. Total RNA was isolated from cells using TRIZOL reagent as per the manufacturer’s instructions. RNA was quantified and an equivalent amount (1 μg) of RNA from each sample was reverse transcribed by using RTase Kit for cDNA synthesis (SuperScript III kit, Thermo Fisher). The cDNA obtained was subjected to quantitative PCR amplification of target-specific genes in an ABI PRISM 7500 qPCR machine. An internal control of 18S rRNA was used for the normalization of target genes in the test conditions. The relative quantification of data from samples was represented as fold change of target genes in samples as compared to control.
Statistical analysis
The data are represented as mean ± standard deviation (SD) from minimal three independent experimental repeats. The data were analyzed by One-Way ANOVA for statistical significance followed by Dunnett’s post-hoc test for multiple comparison of parameters among the groups. p values <0.05 were considered statistically significant.
Results
Phytic acid suppresses proliferation of CRC cells
PYT has exhibited potent anti-inflammatory, cytotoxic, and antioxidant effects in different acute and chronic conditions. Thus, this study explored the antiproliferative effects of PYT on CRC cells HCT116 and HT-29 and deciphered the prospective molecular mechanism involving NF-κB-COX-2-β-catenin signalling pathways. PYT extracted from rice bran was shown to inhibit the growth several cancers cell (breast, liver, and ovary) with an average 50% growth inhibition concentration (IC50) values ranging between 1.6 to 3.7 mm. 26 PYT analogue IP6 was reported to suppress the proliferation of breast cancer cells in a synergistic manner when cotreated with adriamycin and tamoxifen, with IC50 values ranging from 1 to 5 mm in MCF-7 and MDA-MB 231 cell lines. 32 In an interesting manner, PYT showed no sensitivity toward a normal NIH 3T3 murine cell line which was similarly observed in this study. PYT showed lesser cytotoxicity against normal human embryonic kidney 293 (HEK 293) cell lines with no significant sensitivity up to 4 mm PYT, while 16% growth inhibition at 6 mm PYT which is a very high pharmacological concentration yet statistically not significant (Supplementary Figure 1).
PYT-treated HCT116 and HT-29 cells were assayed for cell viability in a time- and dose-dependent manner. Results show that PYT inhibited cell viability in both cell lines in a dose-dependent manner (Figure 1(a) and (b)). PYT caused a prominent reduction in cell viability at 2 mm PYT in both cell lines and both cell lines showed about 50% cell viability as compared to control (100%) at 3 mm PYT. This cytotoxic profiling of CRC cells treated with PYT was utilized to measure IC50 values. Results show that IC50 values estimated for PYT on HCT116 and HT-29 cells were 2.96 and 3.35 mm, respectively. In addition, a time-course study was performed with both cell lines at 2.5 mm of PYT at different hours of incubation. Results show that PYT inhibited the growth of HCT116 and HT-29 cells both in a time-dependent manner with statistically significant growth inhibition at 24 h (Figure 1(c)). We also visualized the cells treated with PYT under a light microscope which showed that HCT116 cells were more responsive and had less cell number at 3.5 mm of PYT. While HT-29 cells were also responsive and showed potent contact inhibition at 3.5 mm of PYT (Figure 1(d)). Furthermore, the clonogenic assay could provide an assessment of the effect of PYT on CRC cell survival and proliferation (Figure 1(e)) which showed that PYT suppressed the colony formation abilities of HCT116 and HT-29 cell lines in a concentration (2.5 and 3.5 mm) dependent manner. Quantification evaluation of clonogenic assay data demonstrated that PYT suppressed the proliferation of HCT116 cells by 62 and 38% at 2.5- and 3.5- mm concentrations, respectively. While the quantitative data of HT-29 cells showed 68 and 42% inhibition of colony formation at 2.5 and 3.5 mm of PYT, respectively (Figure 1(f)). Comparatively HCT116 cells showed notable time-course and dose-dependent effects as compared to HT-29 cells. These observations established that PYT has an antiproliferation effect on CRC cells. Effect of phytic acid on viability of CRC cell lines. HCT116 (a) and HT-29 cells (b) were treated with PYT at indicated conc. For 48 h and dose-dependent cell viability was assessed. (c) HCT116 and HT-29 cells were treated with PYT (3 mm) and time-dependent cell viability was assessed. Percentage of viable cells was presented against vehicle-treated control (100% viability). Data was presented as mean ± SD (d) HCT116 and HT-29 cells were treated with PYT at 2.5- and 3.5-mM conc. For 48 h and then cells imaging was performed under a light microscope at magnification 10×. (e) HCT116 and HT-29 cells were treated with PYT at 2.5- and 3.5-mm conc. Followed by clonogenic assay and (f) quantitative counting of colony forming cells. PYT, phytic acid; *p < 0.05 vs control.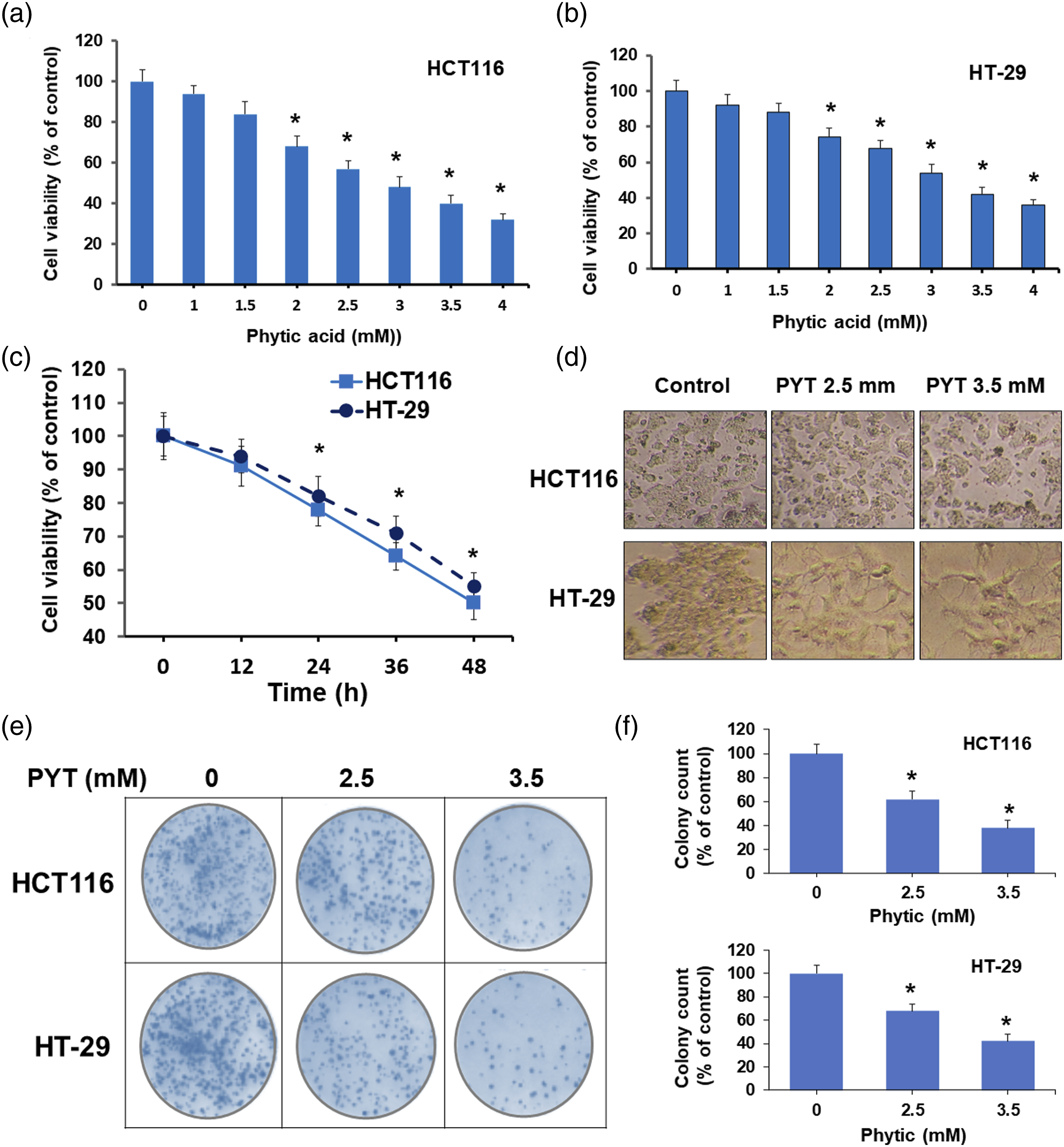
Phytic acid induced apoptosis and cell cycle arrest in colorectal cancer cells
In order to understand the antiproliferation effects of PYT, we assessed its effects on the processes of apoptosis induction and cell cycle by analyzing the level of DNA fragmentation, mitochondrial membrane potential, flow cytometry for apoptosis and cell cycle progression, and Western blotting of apoptosis-related proteins. PYT treated HCT116 and HT-29 both cell lines showed induction of apoptosis in a concentration (2.5 and 3.5 mm) dependent manner (Figure 2). Apoptosis assay by flow cytometry (Figure 2(a) and (b)) and quantitative expression of data (three experimental repeats) showed that PYT (2.5 mm) treatment to HCT116 cells caused the movement of 22.3 ± 3.5% of cells to apoptosis (early and late apoptosis). While PYT (3.5 mm) treated HCT116 cells showed the movement of 34 ± 5.2% of cells to apoptosis (early and late apoptosis). PYT (3.5 mm) treated HCT116 cells also showed 5.3 ± 1.5% cells in the necrosis phase. Likewise, PYT-treated HT-29 cells showed apoptosis induction yet with a slightly different pattern. PYT (2.5 mm) treated HT-29 cells showed 22 ± 4.0% cells in apoptosis phases (early and late apoptosis). While PYT (3.5 mm) treated HT-29 cells showed a total of 30 ± 7.0% cells in apoptosis phases (early and late apoptosis). Mitochondrial membrane potential (ΔψM or MMP) serves as an important molecular biochemical parameter for mitochondrial activity and integrity and thus an indicator of normal or apoptotic cell. The MMP in cells was estimated by using JC-10 staining which is a lipophilic cationic dye that forms a red fluorescent aggregate in normal functioning cells. Whereas red fluorescence was reduced because of diffusion of JC-10 monomeric form in apoptotic or necrotic cells. Therefore, normal functioning cells showed a higher ΔψM, and apoptotic cells showed a lower ΔψM due to JC-10 monomeric form with green fluorescence. HCT16 and HT-29 cells were treated with PYT (2.5 and 3.5 mm) and JC-10 fluorescence was estimated by ELISA analysis (Figure 2(c)). Both the HCT116 and HT-29 cell lines treated with PYT showed a concentration (2.5 and 3.5 mm) dependent reduction in JC-10 fluorescence. As compared to control (100%) HCT-116 cells, PYT-treated cells showed 78 and 62% relative JC-10 fluorescence level at 2.5 and 3.5 mm, respectively. Similarly, HT-29 cells showed 82 and 66% JC-10 relative fluorescence levels at 2.5 and 3.5 mm respectively as compared to control (100%). Reduced ΔψM directly correlates with the disorganization of the mitochondrial membrane which causes the initiation of the apoptotic cascade. Apoptosis once induced causes degradation of the nuclear membrane and fragmentation of genomic contents. Initiation of apoptotic causes activation of intracellular nucleases that cleave large chromatin structural domains into smaller fragments of size 50–300 kb which are further fragmented into smaller pieces of DNA to about 200 bp in size. Thus, the effect of PYT on apoptotic process was confirmed by analysing the internucleosomal DNA fragmentation in HCT116 and HT-29 cells by separating ethidium bromide-stained genomic DNA on agarose gel. Results showed that PYT caused chromatin breakdown in PYT concentration (2.5 and 3.5 mm) dependent manner in both cell lines with a clear DNA banding pattern of about 200 bp (Figure 2(d)). Effect of phytic acid on apoptosis and cell cycle in CRC cells. (a) PYT at 2.5- and 3.5-mm conc. For 48 h HCT116 and HT-29 cells were treated with PYT at 2.5- and 3.5-mm conc. And stained with annexin V/PI followed by flow cytometric analysis for apoptosis. The left-below gate shows normal cells; right-below gate shows early apoptotic cells; right-upper gate shows late apoptotic cells; left-upper gate shows the necrotic cells. (b) Mean of the percentage of cells population in different phases of cell proliferation, normal and apoptosis (early and late apoptosis), was represented as quantitative bar graph. (c) HCT116 and HT-29 cells were treated with PYT at 2.5- and 3.5-mm conc., genomic DNA was isolation and separated on agarose gel electrophoresis for DNA fragmentation analysis. (d) HCT116 and HT-29 cells treated with PYT at 2.5- and 3.5-mm conc. And then followed by JC-10 staining and fluorescence measurement. (e) HCT116 and HT-29 cells were treated with PYT at 2.5- and 3.5-mm conc. Followed by PI staining and flow cytometric analysis for cell cycle progression. (f) The mean of the percentage of cells in different stages of cell cycle in was represented as quantitative bar graph. PYT, phytic acid; PI, propidium iodide.
Additionally, we examined the effect of PYT on cell cycle progression in CRC cell lines by flow cytometry. PYT caused a dose-dependent modulation of cell cycle progression and checkpoint arrest. Results show that PYT caused a reduction of cells in the G phase followed by stagnant accumulation in the S phase and a notable increase of cells in the G2/M phase (Figure 2(e) and (f)). PYT caused accumulation of HCT116 cells in the G2/M phase by 11, 21, and 29% at 0, 2.5, and 3.5 mm PYT, respectively. Furthermore, PYT caused a reduction of cells in G1 phase by 60, 49 and 39% at 0, 2.5 and 3.5 mm PYT, respectively. Likewise, in HT-29 cells, PYT caused a decrease of cells in the G phase by 57, 46, and 37% at 0, 2.5, and 3.5 mm PYT, respectively. While PYT caused accumulation of cells in the G2/M phase by 12, 19, and 28% at 0, 2.5, and 3.5 mm PYT, respectively. These results of cell cycle analysis suggest that PYT caused a G2/M phase cell cycle arrest in CRC cell lines and that cell cycle arrest may be one of the mechanisms behind the antiproliferative effects of PYT on CRC cells.
Results showing that PYT induced apoptosis and caused cell cycle arrest in HCT116 and HT-29 cells were further confirmed by analyzing the protein levels by Western blotting (Figure 3). Bid is a pro-apoptotic member of the Bcl-2 protein family proteins which is known to initiate the apoptotic process.
33
PYT (2.5 and 3.5 mm) treatment to HCT116 and HT-29 cells showed a dose-dependent decrease in the level of total Bid protein which indicates its truncation and translocation (Figure 3). Next, we observed that PYT treatment to HCT116 and HT-29 cells caused a reduction in the level of pro-caspase-9 and increase in the level of cleaved-caspase-9 in a dose-dependent (PYT 2.5 and 3.5 mm) manner. Since caspase-9 cleavage is known to lead to activation of executioner caspase-3, a similar observation was noted from blotting results. PYT caused a reduction in the level of pro-caspase-3 and increase in the levels of cleaved-caspase-3 in both cell lines in a dose-dependent manner. The catalytic activity of caspase-3 utilizes cleavage of a nuclear substrate poly (ADP-ribose) polymerase (PARP) which actually initiates nuclear instabilities as apoptotic marker.
34
PYT treatment to both cells further caused cleavage activation of PARP in a dose-dependent manner. Collectively these results suggest that PYT induced mitochondrial intrinsic apoptosis in CRC cell lines through caspase-3 dependent mechanism. These observations add to that apoptosis induction is one of the mechanisms behind the antiproliferation effects of PYT on human CRC cells. Effect of phytic acid on apoptosis cascade in CRC cells. HCT116 and HT-29 cells were treated with PYT at 2.5- and 3.5-mm conc. Followed by total protein isolation and Western blotting for proteins in apoptosis cascade.
Phytic acid downregulates NF-κB and β-catenin signalling in colorectal cancer cells
To elucidate the intervening mechanism behind the effects of PYT on the proliferation of CRC cells, the role of NF-κB and β-catenin signalling pathways was studied as lead targets. NF-κB/Rel family contains five protein members named as RelA (p65), c-Rel, RelB, NF-κB1 (p105/p50), and NF-κB2 (p100/p52). We studied the effect of PYT on the expression of NF-κB (p65) protein in CRC cell lines by treating HCT116 and HT-29 cells with PYT (2.5 and 3.5 mm) and Western blotting. Results show that PYT caused inhibition of the p65 subunit of NF-κB protein in a dose-dependent manner in both cell lines (Figure 4). PYT also caused inhibition of the phosphorylated form of the p65 (p-p65) subunit of NF-κB in a similar manner in both cell lines. We also assessed the effect of PYT in IκBα (nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha) which is an important protein family member that inhibits the NF-κB transcription factor. IκBα inhibits NF-κB activity by masking its nuclear localization signals and keeping them inactively sequestered in the cytoplasm.
35
Our results showed interesting observations that PYT caused a dramatic increase in IkBα in both cell lines in a dose-dependent manner, especially at 3.5 mm concentration (Figure 4). These results provide further key inputs that PYT not only downregulates the NF-κB levels in CRC cells, but it also limits its phosphorylation activation and associated activities in CRC cells. Inhibition of p65 and p-p65 subunits of NF-kB in CRC cells by PYT was characterized by activation of Bid as apoptosis regulator protein (Figure 3) which in turn correlates with the antiproliferative properties of PYT. These results clearly suggest that PYT inhibits the activity of NF-κB in CRC cells and that would be ultimately regulating the connecting signalling events. Effect of phytic acid on NF-κB and β-catenin pathway proteins in CRC cells. HCT116 and HT-29 cells were treated with PYT at 2.5- and 3.5-mm conc. Followed by total protein isolation and Western blotting for proteins in NF-κB and β-catenin signaling pathway.
The role of Wnt/β-catenin signalling and APC and GSK-3β genes are well documented as crucial factors for colorectal carcinogenesis.16,17 We observed the expression of β-catenin in CRC cells treated with PYT at 2.5 and 3.5 mm by Western blotting (Figure 4). PYT remarkably suppressed the expression of β-catenin in both cell lines in a dose-dependent manner. GSK3β is a negative regulator of β-catenin expression and is directly associated with the development of carcinogenic events in the colon. 16 GSK3 is a serine/threonine kinase which plays a key role in the regulation of the Wnt/β-catenin signalling pathway and thus is a critical protein in the regulation of cell proliferation, differentiation, and maturation. GSK3 phosphorylation triggers β-catenin destabilization. 36 Our results show that PYT caused an increase in the level of GSK-3β in both cell lines in a dose-dependent manner (Figure 4). PYT was further shown to consequently inhibit the levels of c-Myc and cyclin D1 in a dose-dependent manner in both cell lines (Figure 4). These inhibitions are associated with suppressed cell growth and cell cycle arrest, as c-Myc regulates cell proliferation while cyclinD1 maintains cell cycle progression.
Phytic acid suppresses inflammation moderators in colorectal cancer cells
NF-κB signalling is known to regulate multiple molecular steps in cancer as well as it regulates immune and inflammatory responses. Our results showing inhibition of NF-κB led to an assumption that it may provide an anti-inflammatory effect for PYT in CRC cells. TCF4-β-catenin binding was also shown to play a major role in the regulation of Cox-2 expression in CRC HT29 cells. 37 This further led to an assumption that PYT mediated suppression of β-catenin may also cause reduced binding of TCF4-β-catenin complex and may cause inhibition of Cox-2 expression, as a mechanism of antiinflammation. Thus, suppression of NF-κB-COX-2-β-catenin crosstalk is critically associated with the induction of apoptosis and limiting inflammation in CRC cells.
Next, we assessed the effect of PYT on gene expression of key inflammatory mediators COX-2, iNOS, IL-1β, Il-6, NF-κB, and β-catenin in both cell lines by qPCR (Figure 5). Results demonstrate that PYT suppressed the expression of these genes in both HCT116 and HT-29 cells in a dose-dependent manner (Figure 5). PYT repressed the expression of COX-2 to 0.82- and 0.68-fold as compared to control (one-fold) and 2.5 and 3.5 mm, respectively. Likewise, PYT suppressed the expression of iNOS to 0.72- and 0.60-fold as compared to control (one-fold) and 2.5 and 3.5 mm, respectively. The expression of other genes was suppressed in a similar manner. Results showed that PYT suppressed the expression of NF-κB gene also to 0.88- and 0.72-fold at 2.5 and 3.5 mm respectively, which demonstrated that PYT suppressed the level of the NF-κB protein as well as its gene levels. Chronic inflammation in CRC features overproduction of pro-inflammatory cytokines such as IL-6, IL-1β, IL-17 and TNF-α. Collectively, these results confirmed the anti-inflammatory effects of PYT along with inhibition of NF-κB and β-catenin signaling towards growth inhibition of CRC cells. Thus, downregulating NF-κB led to the inhibition of inflammation by PYT appears to be a characteristic plausible mechanism responsible for inhibiting the proliferation of CRC. Effect of phytic acid on inflammation mediators in CRC cells. HCT116 and HT-29 cells were treated with PYT at 2.5- and 3.5-mm conc. For 24 h. RNA was isolated followed by cDNA synthesis and quantitative real-time PCR for specific genes in inflammation pathway.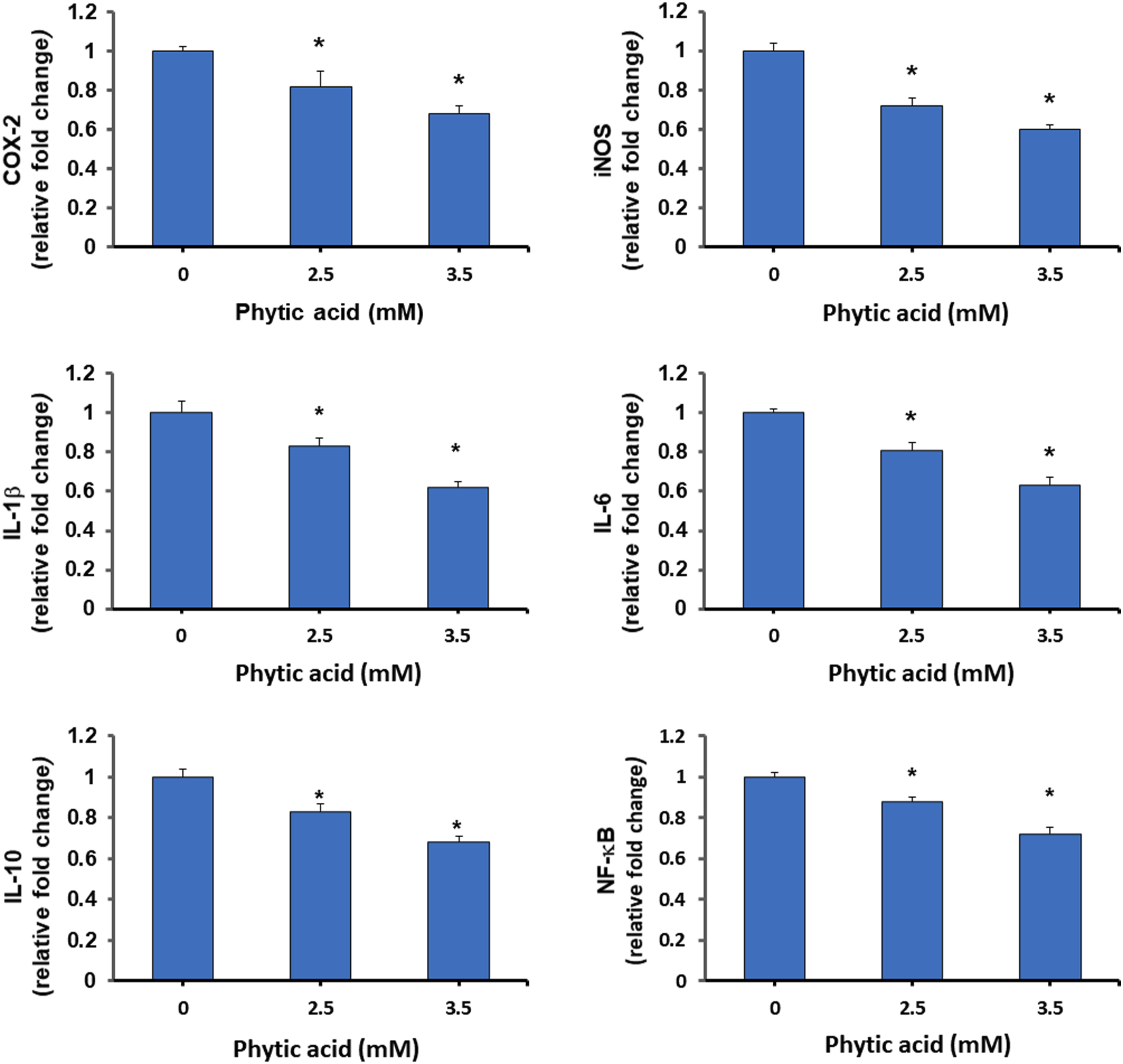
Discussion
Colorectal carcinogenesis involves critical impart of chronic inflammation as multistep molecular event which include intestinal immune system dysfunction, overproductions of cytokines and chemokines. Although the molecular mechanisms of inflammation-driven carcinogenesis are still under exploration, yet there are several key signaling pathways actively engaged. Primarily, the Wnt/β-catenin signaling pathway has been reasonably overactivated in various human cancers mainly colorectal cancers as well as lung, prostate, breast and liver cancer.38–42 Secondly, the NF-κB pathway plays key roles in regulating inflammation and immune response in pathologic conditions. NF-κB pathway governs multiple mechanisms in colorectal carcinogenesis and tumor survival through inhibition of apoptotic cellular pathways, induction of proliferation, and controlling the expression of genes involved in immune and inflammatory function. 8 The proliferation of CRC cells towards tumorigenic development is recurrently associated with mutations in cancer related genes and epigenetic silencing of tumor suppressor genes.43,44
Based on the available literature with growing evidence suggest that PYT has proven records of improving dietary physiology and prevention of pathophysiological conditions including different cancers.12,22–25 Cancer cells have shown diminished apoptosis and high cellular proliferation which is executed in two different pathways, caspase-9 dependent mitochondrial intrinsic and caspase-8 dependent extrinsic.45,46 Thus, reactivating apoptosis in cancer cells is a key approach to limit their uncontrolled proliferation. The cytotoxic profiling of PYT-treated CRC cells showed a time- and dose-dependent response and the estimated IC50 values for HCT116 and HT-29 cells were 2.96 and 3.35 mm, respectively. These cytotoxicity profiles of PYT are comparatively similar to those reported for ovarian, breast, and liver cancers which were in the range of 2–5 mm.26,32 Further the application of PYT on CRC cells showed that it induced apoptosis in both cell lines in a dose-dependent manner with a greater number of cells moving towards late apoptosis phase than early apoptosis. Apoptosis induction is usually correlated with the disturbed mitochondrial membrane functionality, as it is noteworthy that apoptosis in cells is initiated through mitochondria which has a certain mitochondrial membrane potential designated to as ΔψM or MMP. 47 PYT caused a potent reduction in the MMP in both cell lines. Apoptosis induction by PYT was also affirmed by DNA fragmentation, which was potentially increased in both cell lines, as an indicator of nuclear disorganization. The cell cycle analysis suggested that PYT caused a G2/M phase cell cycle arrest in both cell lines which may be a potential mechanism of action to suppress the growth and proliferation of CRC cells. The mechanism behind apoptosis inducing effects of PYT were further confirmed by analysis of apoptosis related protein levels. PYT treatment to caused dose-dependent decrease in the level of total Bid protein which is a pro-apoptotic member of the Bcl-2 protein family that gets truncated (t-Bid) and translocates into the mitochondria and initiates the apoptotic process. 33 Bid truncation and translocation is further known to induced cytochrome c release from mitochondria and activate apoptotic cascade. 48 Recently a sesquiterpene cedrol was shown to activate Bid and initiate apoptosis by activating caspases (9 and 3) in human leukaemia and colon cancer cells. 49 PYT further caused activation of caspase-9 and caspase-3 in both cell lines followed by catalytic cleavage of PARP which collectively emphasizes the apoptotic process in cells. 34
In order understand the mechanism pertaining behind the effects of PYT on the proliferation of CRC cells, the role of NF-κB and β-catenin signalling pathways was explored. PYT was found to inhibit the expression of NF-κB (p65) protein as well as the phosphorylated form of the p65 subunit of NF-κB (p-p65) in both cell lines. Whereas PYT enhanced the expression of IκBα which is an important negative regulator of NF-κB which acts by masking its nuclear localization. 35 Furthermore, the role of Wnt/β-catenin signalling was explored as they act as a crucial factor for colorectal carcinogenesis.16,17 PYT caused a dramatic suppression in the expression of β-catenin in CRC cell lines and enhanced the expression of GSK3β which is a negative regulator of β-catenin. 16 GSK3 is a serine/threonine kinase which plays a key role in the regulation of the Wnt/β-catenin signalling pathway and thus is a critical protein in the regulation of cell proliferation, differentiation, and maturation. GSK3 phosphorylation triggers β-catenin destabilization. 36 β-Catenin is known to form a nuclear complex with the transcription factor T cell factor/lymphoid enhancer factor (TCF/LEF) and controls the expression of numerous genes that regulate cell growth and proliferation such as cyclin-D1, c-Myc, and PCNA.16,17 Our results showed that PYT consequently inhibited the levels of cyclin D1 and c-Myc in CRC cell lines. NF-κB signalling regulates multiple molecular steps in carcinogenesis and tumor survival by suppressing apoptosis, activating cell proliferation, and regulating the expression of genes associated with immune and inflammatory responses.2,3,8,9 NF-κB acts as a transcriptional upregulator of COX-2 and thus affects the crosstalk with iNOS which works coherently with COX-2. NF-κB downregulation in turn associated with the inhibition of cell proliferation and activation of the apoptotic cascade as shown by sensitizing colon cancer cells with TNF-α. 8 PYT was found to notably suppress the levels of iNOS and Cox-2 genes in CRC cell lines. A natural flavonoid fisetin was reported to induce apoptosis in CRC cells by inhibiting NF-κB and Wnt/EGFR signalling pathways and regulate inflammation by inhibiting Cox-2. 38 We have further observed that the expression of the IL1β, IL-6, IL-10 and NF-κB was notably suppressed by PYT treatment. The hyper-activation of immune cells causes overproduction of pro-inflammatory cytokines and chemokines that mainly included overexpression of IL-1β, IL-6, TNF-α, and IFN-γ which are often observed as mediators of intestinal inflammation in IBD patients and as well as CRC in animal models.50,51 While inhibition of IL-1β and IL-6 was found to be highly effective in controlling inflammation responses in the colon. 52 These cytokines have an interesting direct regulatory impact on the NF-κB pathway via creation of a positive autoregulatory loop. 3 Summarily this study attempted to establish that phytic acid suppressed proliferation of CRC cells and its inflammatory state by modulating the expression of β-catenin and NF-κB signalling pathways. Results of the study are well descriptive and conclusive yet to mention the limitation of the study, further experiments may be conducted on in vivo animal model for CRC. This may provide extensive molecular understanding for the use of phytic acid as a supplement for managing inflammatory conditions as complementary and alternative medicine as it abundant in several edible seeds, legumes, nuts, and whole cereals.
Conclusion
Colorectal carcinoma is resultant of alterations in a series of molecular steps including chronic inflammation and growth regulation. Extensive research are conducted on using natural products and single molecules for therapy and prevention of CRC. Natural product-derived small molecule inhibitors acting as antiproliferative, and anti-inflammatory agents have been extensively explored as a therapeutic strategy against CRC. Phytic acid or IP6 obtained from several kinds of cereals, legumes, nut oil, seeds, and soybeans have been reported as potential inhibitors of colon cancer growth. This study explored the effect of PYT on the growth of colorectal cancer cells HCT116 and HT-29 and analysed the involvement of NF-κB and β-catenin signalling. PYT showed dose-dependent growth inhibition of HCT116 and HT-29 cells with an estimated IC50 value of 2.96 and 3.35 mm. PYT induced mitochondrial intrinsic apoptosis pathway in both cell lines by activating caspase-9 and caspase-3, as well as it induced a G2/M phase cell cycle arrest. PYT was shown to suppress the expression of NF-κB/p65 and p-p65 along with activation of IkBα. PYT was further found to interfere with the activity of β-catenin at transcriptional and translational levels as well as activation of GSK-3β. PYT was found to suppress the levels of cMyc and Cyclin D1, the two major proteins involved in cell proliferation and cell cycle progression. PYT was further found to suppress the expression of proinflammatory cytokines and chemokines such as COX-2, iNOS, IL-1β, IL-6, and IL-10 in CRC cell lines. Collectively these observations suggest that PYT suppressed the proliferation of CRC cells by inducing mitochondrial intrinsic apoptosis and suppression of inflammation modulation of NF-κB and β-catenin signalling pathways. To mention as the limitation of the study may be its unexplored mechanism in animal models which may be further studied to add to the repertoire for utilizing phytic acid as a natural complementary and alternative supplement in the prevention of intestinal inflammation and colorectal cancer.
Supplemental Material
Supplemental Material - Phytic acid regulates proliferation of colorectal cancer cells by downregulating NF-kB and β-catenin signalling
Supplemental Material for Phytic acid regulates proliferation of colorectal cancer cells by downregulating NF-kB and β-catenin signalling by Jianyu Feng, Yandong Liu, Chunnan Zhang, Menglin Ji and Cuiyun Li in European Journal of Inflammation.
Footnotes
Acknowledgments
Authors are thankful to the department and institute for providing infrastructure and experimental research facilities.
Author contributions
JF and YL contributed to the design, organization, and performance of the whole experiment. CZ and ML contributed to the secondary experiments and statistics and prediction. CL contributed to manuscript preparation and submission.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.