
Abstract
Accumulating evidence suggests that inflammation is involved in the development of late onset Alzheimer’s disease (LOAD). However, it is not clear whether inflammation is a cause or consequence, or both. The aim of this paper is to review the relationship between inflammation and LOAD. We also review the effect of anti-inflammation on the risk of LOAD to further elucidate the relationship between inflammation and LOAD.
Introduction
Traditionally, Alzheimer’s disease (AD) is subdivided into early onset (EOAD) and late onset forms. EOAD (onset before age 65) accounts for 1–5% of total AD and is primarily associated with disease-causing mutations in presenilin 1, presenilin 2, or amyloid precursor protein (APP) genes. 1 In contrast, LOAD (onset above age 65) accounts for over 95% of total AD and is thought to be associated with multiple factors, including genetics, lifestyles, and environment. 2 Numerous studies have suggested that inflammation is involved in the development of LOAD, but it is not clear whether inflammation is a cause or consequence, or both. The aim of this paper is to review the relationship between inflammation and LOAD and provide possible intervention strategies for LOAD prevention.
Well-established risk factors for LOAD and inflammatory phenotype
Aging and inflammatory phenotype.
Aging is a major risk factor for AD. Among young people, AD is rare; however the risk of AD is greatly increased after age 65. As for the relationship between aging and inflammation, there is consistent evidence indicating that chronic low-grade inflammation characterized by raised serum levels of C-reactive protein (CRP) and pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin (IL)-6 is one of the main characteristics of aging. 3 Such aging-related chronic inflammation is also termed inflammaging and tends to be considered as a possible mechanism underlying the aging process.
Genetic risk factors and inflammatory phenotype.
Apolipoprotein E ε4 allele (APOE4) is the strongest genetic risk factor for LOAD, 4 and has been reported to be associated with enhanced inflammatory responses compared with apolipoprotein E ε3 allele (APOE3), the most common allele of APOE gene. In in vitro studies of microglia, it was observed that APOE4 but not APOE3 enhanced innate immune responses of microglia including altered cell morphology, increased NO production, and higher pro-inflammatory cytokine levels, such as TNF-α, interleukin-1beta (IL-1β), and IL-6.5,6 In targeted-replacement mice expressing APOE4 7 and in human subjects carrying APOE4, 8 enhanced innate immune responses were also observed after intravenous lipopolysaccharide (LPS) administration. Moreover, Fan et al. 9 found that the APOE2/4, APOE3/4, and APOE4/4 carriers had increased levels of inflammatory factors such as TNF-α, IL-6, and IL-1β when compared with non-APOE4 carriers as a whole, respectively.
In addition to APOE4, large-scale genome-wide association studies have identified a growing number of genetic variants associated with the risk of LOAD.10,11 Quite a few of them, such as the rs75932628 polymorphism of the triggering receptor expressed on myeloid cell 2 (TREM2) gene, rs6656401 polymorphism of the complement component receptor 1 gene, and rs3865444 polymorphism of CD33 gene, are located at inflammation-related genes, further highlighting the important role of inflammatory pathways on the onset and/or progression of LOAD. Functionally, genetic variants of the inflammatory pathways may affect the inflammatory phenotype. For example, it was observed that pro-inflammatory cytokines and interferon type I response were upregulated in the brain of TREM2 rs75932628 carriers who developed AD. 12
Environmental risk factors for LOAD and inflammatory phenotype.
Infection with certain pathogens such as spirochetal bacteria and periodontal pathogens has been identified to be a potential risk factor for LOAD.13-15 With regard to the relationship between infection and inflammation, it is known that infection can trigger an inflammatory reaction. Typically, infection-triggered inflammatory reaction is transient and acute; however, it can become chronic and harmful when pathogens evade destruction by the host immune system.
In addition to certain pathogens, emerging evidence from animal and clinical studies suggests that gut microbiota may also play an important role in the development of AD.16,17 Regarding the relationship between gut microbiota and inflammation, it has been reported that gut microbiota dysbiosis can upregulate local, systemic, and central nervous system inflammation. For example, the results of Shen et al. 18 and Shukla et al. 19 showed gut microbiota dysbiosis could upregulate the expression of NOD-like receptor protein 3 (NLRP3) in the intestinal tract of AD model mice, subsequently causing the release of inflammatory factors, which in turn increased AD-associated neuroinflammation. LPS from gut microbiota, which can increase the permeability of blood–brain barrier (BBB) and induce systemic inflammation and AD-like pathology, may represent one of the candidate pathophysiologic links between gut microbiota dysbiosis and AD pathology.20,21
Head trauma is also a well-established environmental risk factor for LOAD. 22 Dead cells in the brain with trauma can cause inflammation characterized by the activation of resident cells, migration, and recruitment of peripheral leukocytes, and the release of inflammatory mediators in the injured areas. Evidence obtained from patients with traumatic brain injury and animal models suggested inflammation caused by trauma not only occurred in the injured areas but also disseminated to the remote areas of the brain within days after trauma. 23
Certain diseases increasing LOAD risk and inflammatory phenotype.
Epidemiological findings have shown that certain non-infective diseases, such as cardiovascular diseases, 24 type 2 diabetes mellitus (T2DM), 25 hyperhomocysteinemia (HHcy), 26 and depressive illness, 27 are well-established risk factors for LOAD. Coincidentally, these diseases are all considered to be associated with a state of chronic inflammation. Among them, the cardiovascular diseases have been accepted to be chronic systemic inflammatory diseases, in which both innate and adaptive immunity play important roles. 28 Lowering inflammation alone has been demonstrated to reduce the risk of cardiovascular diseases. The most noteworthy study is the canakinumab anti-inflammatory thrombosis outcome study, showing that therapy with canakinumab, a monoclonal antibody targeting the IL-1β innate immunity pathway, improved the outcome of patients with acute coronary syndromes and evidence of systemic inflammation. 29 As for T2DM, it is also confirmed as a pro-inflammatory state. 30 Compared to normal subjects, patients with T2DM have significantly increased levels of inflammatory markers such as IL-6, IL-8, and TNF-α.31,32 Presently, the mechanisms of inflammation activation in T2DM are largely unexplored. With regard to HHcy which has been thought to have a causal relationship with LOAD, it is actually considered as an inducer of inflammation and a marker of inflammation. It has been shown that HHcy promotes the production of pro-inflammatory cytokines such as IL-1β, IL-6, and TNF- α in different tissues and cells.33,34 Activation of NLRP3 inflammasomes may contribute to HHcy-induced inflammation. 35 Depression is also a well-established risk factor for LOAD. Accumulating clinical and preclinical evidence suggests that chronic low-grade inflammation plays a significant role in the pathophysiology of depression. 36 It has been observed that in the peripheral blood of patients with depression, levels of inflammatory markers such as IL-1β, TNF-α, and CRP are significantly elevated. 37 Furthermore, anti-inflammatory interventions have shown antidepressant effects, making chronic low-grade inflammation increasingly become a therapeutic target for depression.36,38
Lifestyle risk factors for LOAD and inflammatory phenotype.
Similar to other risk factors for LOAD, lifestyle risk factors such as obesity 39 and physical inactivity 40 have also been reported to be closely associated with a state of chronic systemic inflammation. Researches have shown that obesity can induce adipose tissue inflammation, which in turn promotes systemic low-grade inflammation leading to many of its complications. 41 Physical inactivity is also associated with higher levels of markers of chronic systemic inflammation, such as IL-6 and CRP. 42
Relationship between β-amyloid (Aβ) pathological hallmark and inflammation
Aβ production is induced under inflammatory conditions.
Aβ deposition in the brain is one of the characteristic pathological markers of AD. Studies have shown that Aβ is induced under inflammatory conditions.
Pathogen infection is an important inflammatory condition that can induce Aβ production. In in vitro experiments, it was observed that exposing mammalian glial or neuronal cells to pathogen such as Borrelia burgdorferi 43 and periodontal pathogen 44 led to Aβ production. In in vivo experiments, it was also seen that infection by a pathogen such as HSV-1 45 and periodontal pathogens 46 resulted in an accumulation of Aβ and AD-like neuronal degeneration in the brains of experimental animals.
Head trauma is another important inflammatory condition that can induce Aβ production. Olsson et al. 47 reported Aβ levels increased up to 1173% from day 0–1 to day 5–6 in the cerebrospinal fluid of patients after severe brain injury and remained elevated for some time after the initial injury event.
Pro-inflammatory mediators can also induce Aβ production. In human neuronal and extraneuronal cells, it was observed that treatment with interferon-gamma (IFN-ϒ) plus TNF-α or IFN-ϒ plus IL-1β triggered Aβ production by promoting beta-secretase cleavage of the immature APP molecule. 48 Presumably it is through inducing the production of pro-inflammatory mediators that infection and head trauma promote the production of Aβ.
Aβ triggers and magnifies inflammatory reaction.
Although evidence indicates that Aβ is an antimicrobial peptide, a class of innate immune defense molecule that utilizes fibrillation to protect the host from a wide range of infectious agents,49,50 excessive Aβ can trigger an inflammatory reaction and magnify pre-existing inflammation in the brain, leading to a vicious cycle of inflammation amplification and Aβ production, which may eventually trigger neuronal dysfunction and loss of function (Figure 1).
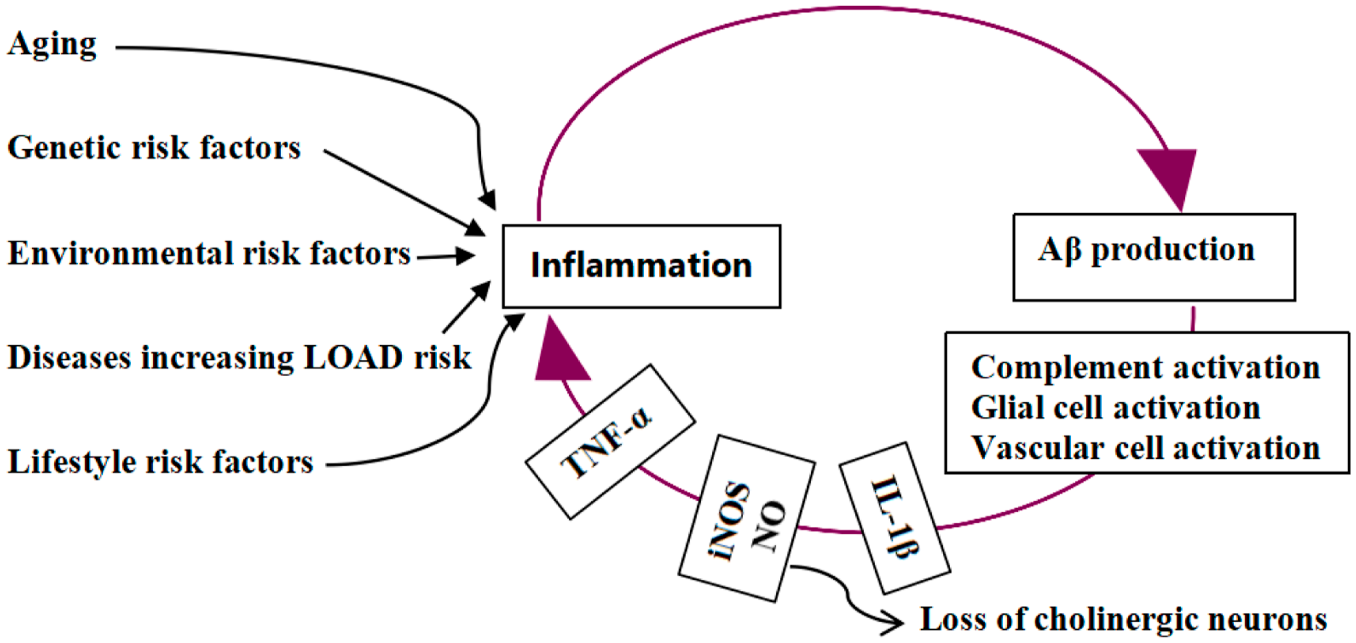
Inflammation and LOAD. Risk factors for LOAD show a common chronic inflammatory phenotype. Aβ production is induced under inflammatory conditions. By activating complement, glial, and vascular cells, Aβ induced by inflammation in return initiates inflammatory reactions and magnifies pre-existing inflammation, leading to a vicious cycle of inflammation amplification and Aβ production and loss of cholinergic neurons.
Data have shown Aβ can trigger and magnify inflammatory reactions by activating microglia, astrocytes, and complement molecules in the brain (Figure 1). Early findings have revealed that Aβ can activate microglia and astrocytes, causing inflammatory responses such as IL-1β production and increased expression of cyclooxygenase and inducible nitric oxide synthase (iNOS) to kill neurons. 51 Recent in vivo studies of AD patients also indicated that Aβ pathology was closely associated with astrocyte activation biomarkers such as chitinase-3-like protein 1 (YKL-40) 52 and glial fibrillary acidic protein (GFAP), 53 indirectly supporting the association between Aβ and astrocyte activation during AD development. Of note, Aβ needs a pre-stimulated environment to show its maximal pro-inflammatory potential. 54 Besides, Aβ can activate complement, an integral component of Aβ plaques in the brain of AD patients, which further exaggerates the neuroinflammatory response induced by Aβ in microglia. 55 Moreover, APOE4 can enhance Aβ-triggered complement activation, reflecting a possible synergistic effect between Aβ and APOE4 in complement activation and neuroinflammation triggering. 56
Studies have indicated that Aβ can also trigger inflammatory reactions by activating vascular cells (Figure 1). Amyloid angiopathy with vascular damage and inflammatory changes is another hallmark in the brains of most AD victims. Growing evidence supports the central role of Aβ in amyloid angiopathy. In cultured human vascular cells, it was observed that Aβ triggered an inflammatory cascade reaction by increasing the expression of inflammatory factors such as IL-1β, IL-6, and IFN-ϒ. 57 In in vivo experiments, Aβ-induced vascular damage and leukocyte activation were also observed using intravital microscopy. 58 Additionally, it was reported that fibrillary Aβ could induce inflammation in the BBB, causing a loss of barrier integrity. 59
It is worth mentioning that Aβ from the peripheral tissue may also have a potential to induce AD pathologies. A parabiosis experiment by Bu et al. 60 showed that human Aβ originated from AD model mice could enter the circulation, accumulate in the brains of wild-type mice, and induce AD-type pathologies including neuroinflammation, tau hyperphosphorylation, and neurodegeneration after a 12-month period of parabiosis. It is noteworthy that platelets, which express large amounts of APP, are a major source of peripheral Aβ.61,62 Researchers have been increasingly interested in investigating the roles of platelet-derived Aβ in AD development. There is evidence showing that activated platelets may contribute to the formation of cerebral amyloid angiopathy63,64 and Aβ deposition in the brain parenchyma. 65 Moreover, in AD patients, increased platelet activation has been observed, 66 which, together with the above evidence, indicates that platelets-derived Aβ may contribute to the development of AD. In future, more evidence needs to be accumulated to deeply elucidate the roles of platelets-derived Aβ in the pathogenesis of AD.
Anti-inflammation shows protective effect on AD
Protective effect of inhibiting TNF-α on AD.
Studies have shown that inhibiting pro-inflammatory mediators has a protective effect on AD. In AD model mice, it was observed that treatment with TNF-α inhibitor XPro1595 before amyloidosis reduced multiple hallmark features of AD, prevented synaptic deficits, and improved memory performance. 67 In humans, Zhou et al. 68 found that the use of TNF-α inhibitor licensed for rheumatoid arthritis (RA) treatment, concurrently lowered the risk of AD among RA patients.
Protective effect of non-steroidal anti-inflammatory drugs (NSAIDs) on AD.
Use of NSAIDs, a class of non-selective inhibitors of cyclooxygenase-1 and cyclooxygenase-2, also presents a protective effect on AD. In AD model mice, when administered early in the disease course, NSAIDs reduced Aβ deposition, tau hyperphosphorylation, and memory impairment. 69 In humans, multiple epidemiological studies have revealed that NSAIDs exposure is associated with a reduced risk of AD, especially in long-term users with a relative risk of around 0.36.70,71
Protective effect of Mediterranean diet (MD) on AD.
MD, a good source of ω-3 fatty acids, polyphenols, probiotics, and vitamins, has anti-inflammatory action. Consistently, results from multiple meta-analyses indicated that higher adherence to MD was associated with a significantly lower risk of AD, 72 reduction in the AD biomarker burden, 73 and decreased risk of conversion from mild cognitive impairment (MCI) to AD, 74 thus making it a very promising approach of AD prevention. 75 Not only that, MD is currently considered as one of the healthiest diets effective against the risk of age-associated diseases. Presently, high-quality randomized controlled trials (RCTs) are still needed to assess the impact of adherence to MD on AD prevention in various populations.
Protective effect of folate on AD.
Folate metabolism is required for several cellular processes including DNA synthesis, repair, and methylation. Folate deficiency leads to HHcy, which is an inducer of inflammation and a well-established risk factor for AD. Consistent results of multiple meta-analyses on the association between folate and AD indicated that folate deficiency increased the risk for AD, while sufficient intake of folate was a protective factor against AD. 76 Furthermore, it was reported that the presence of lower serum folate levels predicted conversion from MCI to all-cause dementia. 74 To further evaluate the prevention performance of folate intake against AD and to determine specific guidelines such as the appropriate age for intervention and duration, high-quality RCTs are still needed in the future.
Protective effect of vitamin D on AD.
In addition to regulating calcium–phosphorus homeostasis, vitamin D also has anti-inflammatory property. The association between serum vitamin D levels and the risk of AD has been widely studied, and most of the meta-analyses suggested that higher levels of vitamin D were associated with a lower risk of AD, making vitamin D supplementation a promising approach to prevent AD.75,77 To further assess the value of vitamin D supplementation in the prevention of AD, and to determine specific guidelines such as the appropriate age for intervention, duration, and amounts, high-quality RCTs are still needed in the future.
Protective effect of anti-inflammatory plant extracts on AD.
In in vivo trials, it has been observed that certain anti-inflammatory plant extracts can attenuate AD pathological deficits through regulation of neuroinflammation.78-80 For example, in a mouse model of AD induced by Aβ1-42, it was observed that intracerebroventricular injection of resveratrol could protect against cognitive decline and significantly ameliorate biochemical changes induced by Aβ1-42. 80 In AD patients, Gu et al. 81 found that resveratrol could attenuate the decline in independent living performance. Regarding the molecular mechanism of resveratrol protecting against AD, it was reported that resveratrol could inhibit Aβ1-42-induced inflammation though nuclear factor kappa-B/NLRP3 signal pathway in glial cells,82,83 and that in human neural stem cells, resveratrol could significantly abrogate Aβ-mediated decrease of cell viability through AMP-activated protein kinase-dependent pathway. 84
Protective effect of probiotics on AD.
Probiotics supplementation, which can decrease inflammation originating from gut microbiota dysbiosis, has been reported to ameliorate AD pathology and cognitive impairment in AD mouse model.85,86 In community-dwelling older adults, it was also observed that probiotics supplementation improved their cognitive function. 87 Generally, evidence of probiotics leading to neurocognitive improvement in humans is still limited; more high-quality observational studies and well-designed RCTs are still needed to evaluate the efficacy of probiotics supplementation on AD prevention.
Protective effect of physical exercise/activity on AD.
Physical exercise/activity, which can reduce inflammation, is gaining more and more evidence as a primary prevention intervention for improving cognitive functions. The effects of physical exercise/activity on the risk of AD and cognitive function have been largely studied. Multiple meta-analyses of epidemiological studies have consistently indicated that physical exercise/activity is a robust protective factor for AD, 88 and a number of meta-analyses of RCTs have suggested that physical activity/exercise positively influences cognitive function in patients with MCI or AD. 89 Results of animal experiments conducted with mice suggested that physical exercise/activity might improve cognitive impairment by augmenting Aβ clearance, reducing Aβ plaques 90 and increasing the levels of peripheral brain-derived neurotrophic factors. 91 To further assess the preventive effect of physical activity/exercise on AD and to establish clear recommendations such as exercise/activity type, frequency, intensity, or duration, more well-designed RCTs are needed in the future.
Conclusion
In summary, several lines of evidence suggest that inflammation may be both a cause and a consequence during LOAD development (Figure 1). Given this, controlling chronic inflammation throughout life may be an appropriate strategy to prevent LOAD. Anti-inflammatory approaches that are easy to implement in daily life and have the potential to prevent LOAD include: (1) higher adherence to MD; (2) sufficient folate intake; (3) vitamin D supplement; (4) probiotics supplementation; (5) supplementation of anti-inflammatory plant extracts such as resveratrol; (6) regular physical exercise/activity. Generally, high-quality prospective studies or RCTs are still needed to determine specific guidelines for these approaches to prevent LOAD.
It should be mentioned that several limitations existed when collecting supporting evidence for the hypothesis that inflammation might be both a cause and a consequence of LOAD. First, when analyzing the relationship between risk factors of AD and inflammatory phenotype, we mainly focused on the well-established risk factors, while those less-established were less analyzed. Second, when analyzing the relationship between pathological markers of AD and inflammation, we mainly focused on the classical markers, while those under clinical validation were less analyzed. Third, when analyzing the protective effects of anti-inflammatory approaches on AD, we tended to focus on the approaches with substantial supporting evidence, while those with less data were less discussed. Finally, the limitation that presently evidence from studies with human subjects is relatively lacking should also be mentioned here. In future, more well-designed studies with human subjects are needed to deeply investigate the action mechanism of inflammation in the development of AD, and to evaluate the primary prevention effect of anti-inflammatory approaches against AD.
Footnotes
Acknowledgements
We thank the reviewers for the critical comments, and thank the journal editorial staff for handling the manuscript.
Author Contributions
Dong Zhao, Jinpei Wang, and Xiaoe Luo participated in the collection of data. Jinrong Zhao and Dong Zhao participated in the data analysis. Jinrong Zhao and Jinpei Wang participated in writing, editing, and revising the manuscript. Rui Guo prepared the figure. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study was funded by the National Natural Science Foundation of China (31100547), Key R & D Program of Shaanxi Province (2022SF-570), the Initiation Funds for High Level Talents Program of Xi’an International University (XAIU202005 and XAIU2019003), and by the Science and Research Special Project of Education Department of Shaanxi Province (19JK0727).