
Abstract
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by immune activation and multi-immunologic phenotypes. Interleukin-33 (IL-33) has been shown to be a critical and pleiotropic immunoregulatory mediator in the pathogenesis of many autoimmune diseases. At present, there are conflicting findings in the research of IL-33 in SLE. The purpose of this study was to investigate whether and how IL-33 is involved in the occurrence and development of SLE.
Methods
43 SLE patients and 43 healthy volunteers were recruited for this study. Serum levels of IL-33, IL-4, IL-6, IL-10 and IL-21 were measured by ELISA. The expression of IL-33 was investigated in kidney sections by immunohistochemistry in lupus nephritis patients (n = 5) and controls (n = 3). The mRNA expressions of Toll like receptor 4 (TLR4), TLR2, and tumorigenicity 2 (ST2)L were quantified in peripheral blood mononuclear cells (PBMCs) by real-time PCR. The surface expression of TLR4 on T cells, B cells, monocytes, and neutrophils was assessed by flow cytometry (n = 22). Mann–Whitney U-test and Spearman’s test were used for statistical analysis.
Results
Serum concentrations of IL-33 were significantly higher in SLE patients than in healthy controls (p < 0.0001). IL-33 expressions were positively correlated with IL-4 and IL-6 levels in SLE patients, which play pivotal roles in the autoantibody production. In addition, TLR4 and TLR2 mRNA were markedly increased in PBMCs from SLE patients (p < 0.05). TLR4 was positively associated with IL-33, while TLR2 was not.
Conclusions
These results imply that upregulated expression of serum IL-33 together with increased TLR4 in PBMCs may contribute to the pathogenesis of SLE via promotion of inflammatory cytokines production.
Introduction
SLE is an autoimmune inflammatory disease characterized by B cell hyperreactivity and the production of anti-nuclear autoantibodies, especially dsDNA and nucleosomes,1,2 resulting in the multi-organ injury including kidney, bone marrow, liver, and so on. The pathogenesis of the disease is complicated and no effective therapy is available. Many cytokines have been demonstrated in the development of SLE, such as IL-4, IL-6, and IL-17.3,4 IL-33, a novel member of the IL-1 cytokine family, has been initially reported as a pleiotropic cytokine that plays a key role of in rheumatic diseases via binding to its receptor ST2. 5 IL-33 favors T helper 2 (Th2) immune response by activating nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK).6,7 High expression level of IL-33 in human rheumatoid arthritis (RA) has been reported, and synovium locally produced IL-33 may contribute to the pathogenesis of joint inflammation and destruction by promoting Th1/Th17 immune response.8,9 IL-33 is also an alarmin and damage-associated molecular pattern (DAMP) molecule. ILC2, Th2, and Tregs are the main target cells of IL-33. In active SLE, the amount of Neutrophil Extracellular Traps (NETs) decorated with bioactive IL-33 was increased, and it was considered to correlate with neutrophil activation, type I IFN production, and end-organ inflammation. 10 Elevated serum IL-33 levels were found in SLE patients.11,12 Given the complexity and functional diversity of IL-33, it is necessary to further investigate its mechanism on chronic inflammatory disorder in SLE.
TLRs broadly distributed on immunocytes are arguably the best-studied immune sensors of invading pathogens and endogenous alarmins. 13 TLRs have also been implicated in several immune-mediated and inflammatory diseases. A wealth of in vitro studies has shown that both autoreactive B cells and plasmacytoid dendritic cells can be activated by TLR ligands. TLR4 inhibitor has been proved to attenuated autoantibody production and renal injury in lupus mice.14,15 TLR4−/− mice demonstrated a global decrease in both Th1 and Th17 associated cytokine production, and the levels of autoantibody and renal injury were also reduced in TLR4−/− mice.16,17 These results demonstrate that TLR4 plays a key role in autoimmunity and organ involvement.
Indeed, previous studies have been shown that TLR4 function was modulated by IL-33 in vitro and sepsis, 18 whereas the relationship between IL-33 and TLR4 has not been described in SLE patients. In the current study, we hypothesized that IL-33 could favor IL-4 and IL-6 cytokines production during the development of SLE through upregulating the TLR4 expression. To address this question, we collected the SLE patients’ sera. Our data demonstrated that The IL-33 expression was considerably elevated in the serum of SLE patients compared with healthy controls. Additionally, a positive correlation of IL-33 with inflammatory cytokines IL-4 and IL-6 was observed, which acts critical role in the autoantibody production. Furthermore, an increased expression of TLR4 in PBMC from patients with SLE showed a significantly positive correlation with IL-33. These results suggest that upregulated expression of serum IL-33 together with increased expression of TLR4 in PBMCs may contribute to the pathogenesis of SLE by promoting inflammatory cytokines production.
Materials and methods
Patients and controls
Demographic and clinical characteristics of the systemic lupus erythematosus patients (n = 43).
Immunohistochemical analysis for IL-33
The anti-IL-33 was obtained from R&D (Minneapolis, MN, US). After deparaffinization and rehydration, the kidney tissue sections were treated with 3% H2O2 followed by blocking with 10% goat serum in PBS. The sections were then stained with anti-IL-33 antibody overnight at 4∘C, and were then used hypersensitive two-step immunohistochemical detection reagent (ZSGB-BIO, China) to detect the IL-33 expression levels by microscope.
Quantification of transcripts by RT-PCR
Total RNA was isolated from PBMCs by Trizol reagent (Invitrogen, USA) following the manufacturer’s instructions. Briefly, 1ug total RNA was first reverse-transcribed to cDNA with reverse transcription reagent kits according to the manufacturer’s protocol (Roche, CH). The expression levels of TLR4, TLR2, and ST2L mRNA were determined by real-time quantitative PCR, using SYBR Green (Roche, Switzerland). Quantitative PCR was performed according to the manufacturer’s instructions (ABI7500, USA). Data was normalized to the GAPDH mRNA expression levels, and the formula 2–ΔΔCt was used to calculate the relative expression. The primers used were as follows: GAPDH, sense primer 5’- GTG AAC CAT GAG AAG TAT GAC AAC -3’, anti-sense primer 5’- CAT GAG TCC TTC CAC GAT ACC -3’; TLR4, Sense primer 5’- GTT GAT GTG GAG AAG GTG TCT G -3’, anti-sense primer 5’- GTA GGC AGG TAG GCA GGG T -3’; TLR2, sense primer TGC TGC CAT TCT CAT TCT TCT G, anti-sense primer AGG TCT TGG TGT TCA TTA TCT TCC; ST2L, sense primer AAC GAG TTA CCA ATA CTT GCT C, anti-sense primer CAG GCA CTA TTG CTT CTG GG.
ELISA analysis of cytokine expressions
Four milliliters of blood were collected in sterile non-treated tubes and were then centrifuged at 3,000 rpm for 5 min at temperature ambient to obtained serum, which was immediately frozen and stored at −80°C. Serum IL-33, IL-4, IL-6, IL-10, and IL-21 were measured by commercially available ELISA assay kits according to the manufacturer’s directions (IL-33 was purchase from PeproTech, USA, other cytokines were obtained from ebioscience, USA).
Flow cytometry
The PBMCs of SLE patients and healthy controls were incubated with the fluorescent-conjugated monoclonal antibodies in the staining buffer. Conjugated Antibodies against CD14, CD3, CD19, and TLR4 were purchased from Biolegend (San Diego, CA, US).
The neutrophils were gated on SSC and FSC.
Statistical analysis
The statistical significance of the data was performed by PRISM software (GraphPad Software, San Diego, CA, USA). Mann–Whitney U-test was used for unpaired samples. The Spearman correlation coefficient was constructed for the determination of linear relationships. p values <0.05 were considered as statistically significant.
Results
Serum IL-33 expression was upregulated in SLE patients
Serum levels of IL-33 were measured in 43 SLE patients and 43 age- and sex-matched controls. Serum IL-33 levels were significantly higher in SLE patients (164.8 ± 130.6 pg/mL) than in healthy controls (64.61 ± 44.67 pg/mL) (p < 0.0001) (Figure 1(a)). To explore the source of serum IL-33 expression, the PBMCs from patients and healthy controls were subjected to analyze the IL-33 expression. However, we did not detect the IL-33 expression in the PBMCs by RT-PCR. IL-33 can be expressed in many tissues, and kidney is the main involved organ of SLE. Therefore, we analyzed the renal tissues of lupus nephritis (LN) patients and healthy controls. As shown in Figure 1(b), a significantly increased IL-33 expression was observed in peritubular regions in lupus nephritis. IL-33 was highly expressed in SLE patients compared with healthy controls. (a) The expression of IL-33 was analyzed in SLE patients and healthy control (n = 43) by ELISA. Mann–Whitney U-test was conducted to test the statistical significance. p value < 0.05 was accepted as statistically significant. (b) The production of IL-33 in kidney tissue was detected by immunohistochemistry.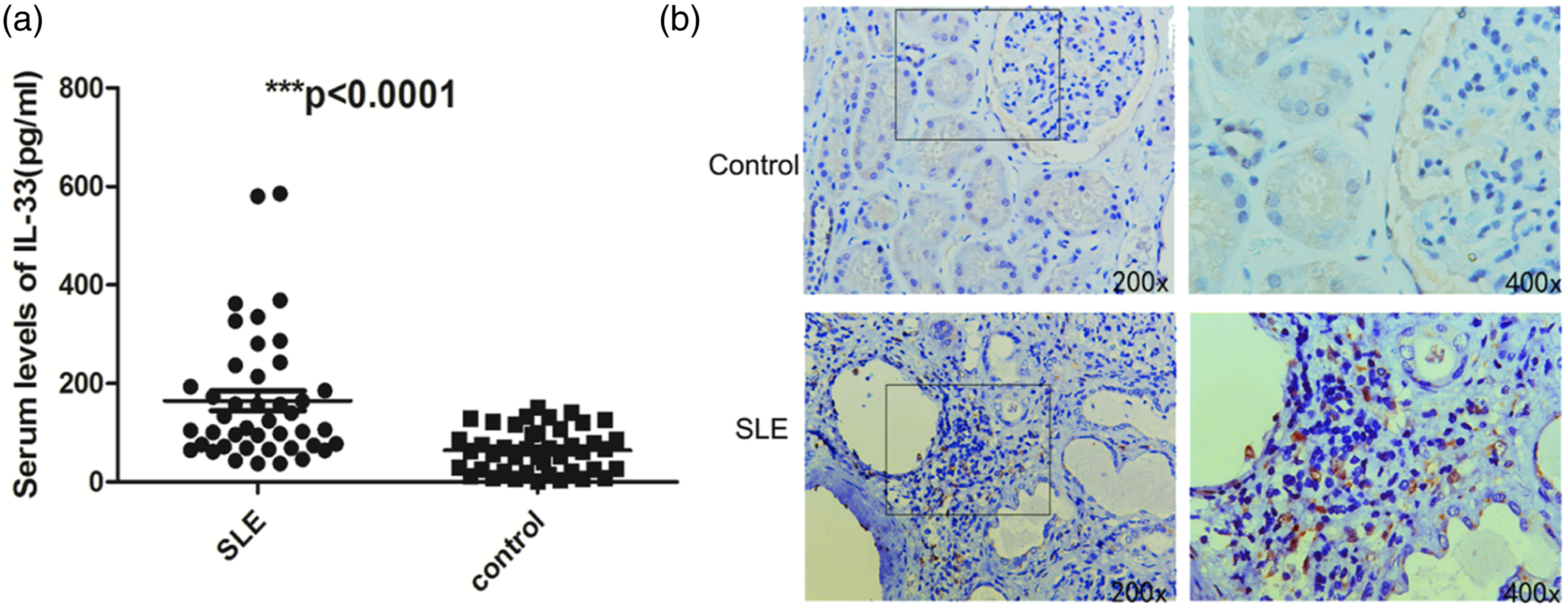
Serum IL-33 level was correlated with inflammatory cytokines levels
Increasing evidence suggests the vital role of IL-33 in the development of SLE,
12
whereas the mechanism remains to be further explored. It was demonstrated that IL-33 can promote inflammatory cytokines production through binding its receptor ST2 expressed in various immune cells.
21
In this study, the correlation between IL-33 and inflammatory cytokines was analyzed. We observed a significantly positive correlation between IL-33 and IL-4 level (r = 0.44, p = 0.003), while increased IL-4 production was implicated in B-cell activation and anti-DNA production in SLE.
22
In addition, IL-6 was also significantly positively correlated with IL-33 (r = 0.41, p = 0.006), which closely link with B lymphocyte maturation into plasma cell and immunoglobulin production in SLE.
23
IL-10 acts as a growth and differentiation factor on cytotoxic lymphocytes and B-cells, and promotes survival of autoreactive B-cells.
24
IL-21 has been considered as a new mediator of inflammation in SLE because of its important role in the growth, survival, differentiation, and function of both T and B cells.
25
However, no significant correlation has been found between their levels and IL-33 level (Figure 2). The correlations of IL-33 with cytokines in SLE patients. (a–d) Sera from the SLE patients were collected (n = 43), then IL-4, IL-6, IL-21, and IL-10 were analyzed by ELISA. Correlations between serum IL-33 expression and cytokines in the SLE patients were analyzed by Spearman’s test. p value < 0.05 was accepted as statistically significant.
Serum IL-33 was positively correlated with TLR4 in PBMC from SLE patients
To explore the mechanism of IL-33 on established proinflammatory cytokines productions, PBMCs were separated from 43 SLE patients and healthy controls. Previous studies have shown that IL-33 could modulate the TLR4 and TLR2 expression,
26
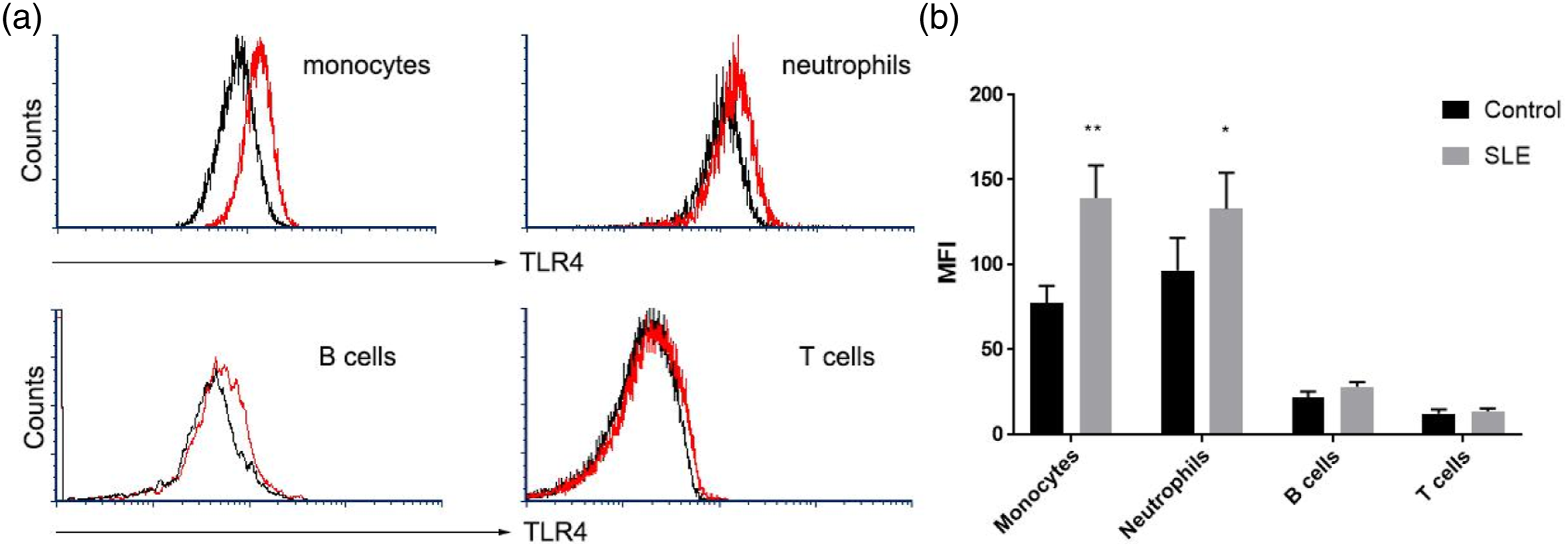
and TLR4/TLR2 plays an important role in the inflammatory cytokines of SLE. To confirm the TLR4/TLR2 regulation by IL-33, quantitative-Real-time PCR was used to analyze the mRNA expression of TLR4, TLR2, and suppression of tumorigenicity 2 ligand (ST2L). Results showed that TLR4 and TLR2 transcripts in SLE patients were significantly higher than those in healthy controls (Figure 3(a)), while ST2L expression was not changed in SLE patients. In addition, we analyzed TLR4 and TLR2 expression in PBMC and serum IL-33. TLR4 was found to be positively correlated with IL-33 in SLE patients (r = 0.37, p = 0.01) (Figure 3(b)), while the correlation between TLR2 and IL-33 was not significant (p>0.05). Specific cell populations expressing TLR4 such as monocytes, granulocytes, and lymphocytes in PBMCs were then identified by flow cytometry. Compared with healthy controls, the TLR4 expression was increased on the monocytes and neutrophils in SLE patients (Figure 4). Serum IL-33 was positively correlated with TLR4 mRNA in PBMCs from SLE patients Expression Profiling of TLR4 on PBMCs from SLE patients. The PBMCs were harvested from the SLE patients and healthy controls for TLR4 expression analysis by flow cytometry (n = 22). (a) The cells were stained with anti-CD3 (T cells), anti-CD19 (B cells), anti-CD14 (monocytes), and anti-TLR4 antibodies. The surface expression of TLR4 on T cells, B cells, monocytes, and neutrophils were plotted in histograms. (b) The TLR4 expressions on monocytes, neutrophils, B cells, and T cells were present by mean fluorescence intensity (MFI). Mann–Whitney U-test was conducted to test the statistical significance. p value < 0.05 was accepted as statistically significant.
Discussion
In the present study, we demonstrated the expression of IL-33 and TLR4 in SLE patients. Current results provided evidences that both IL-33 and TLR4 expression were significantly elevated in this disease. IL-4 and IL-6 levels were shown to be positive correlated with IL-33 expression. Furthermore, a positive correlation between IL-33 in sera and TLR4 mRNA in PBMCs was observed. These data suggest an important role of IL-33 as inflammation promoter in SLE, by upregulating TLR4 expression and resulting in the inflammatory cytokines IL-4 and IL-6 production.
SLE is a multi-systematic disease characterized by chronic immune activation and multiple immunologic phenotypes. Th1, Th17, and Th2 responses have been implicated in the pathogenesis of lupus.27,28 Accumulating evidences show that inflammatory cytokines act a critical role in the development of SLE. 29 These cytokines could promote the T cells and B cells differentiation. IL-33 is a novel member of IL-1 family which was first reported by Schmitz in 2005. 30 Plenty of studies have demonstrated that IL-33 promotes Th2 immune response through ST2L expressed on the Th2 cells. It was shown that IL-33 plays a pathogenic role in asthma through the activation and production of type II cytokines. 31 Furthermore, IL-33 can reduce atherosclerosis and inhibit transplant rejection by converting Th1/Th17 into Th2 immune response,32,33 which is mediated primarily by Th1 and Th17 cells. In our previous study, we also have shown that IL-33 ameliorates inflammatory bowel disease (IBD) partly through promoting Th2 response. 34 There have been multiple evidences confirming the involvement of IL-33 in RA. 35 After that, the high expression of IL-33 in human RA synovium and experimental arthritis have been detected. Accordingly, IL-33 treatment exacerbated the antigen-induced arthritis by activating mast cells. In this study, our data also showed that IL-33 expression was significantly increased in SLE patients compared with healthy controls, which was consistent with previous evidences supporting the vital role of IL-33/ST2 signaling in the pathogenesis of SLE.36,37 Upregulated serum IL-33 level was closely associated with the abnormal changed C-reactive protein (CRP) level and erythrocyte sedimentation rate (ESR). 38 The serum ST2 level of patients with active SLE was significantly higher than that of patients with inactive SLE and normal controls. 39 These results strengthened the idea that IL-33/ST2 signaling may play a crucial role in the acute phase of the disease. In our study, IL-33 was not associated with disease activity or specific organ involvement in SLE patients (data were not shown), which may be due to the relatively small number of active patients enrolled and the fact that IL-33 plays a part role in the pathogenesis of SLE.
For autoimmune diseases, IL-33 expression was altered in the serum of active patients, and this may be correlated with inflammatory cytokines. In the earlier studies, it was reported that administration of sST2 fusion protein, the IL-33 decoy receptor, dramatically attenuated disease severity by inhibiting the release of proinflammatory cytokines comprising IL-6, IL-12, tumor necrosis factor-α (TNF-α), and interferon-γ(IFN-γ). 40 In other rheumatic diseases, such as ankylosing spondylitis, idiopathic inflammatory myopathies, and Behçet’s disease, serum IL-33 levels were positively correlated with inflammatory cytokines IL-13, IL-4, IL-17, and TNF-α levels.31,41 In line with above results, our present study also shows that IL-4 and IL-6 were positively correlated with IL-33 expression in SLE patients. IL-4 production by double-negative T cells has been shown to be increased and related with B-cell activation and anti-DNA production in SLE. Besides the critical cytokine for the Th17 differentiation, IL-6 is one of the first cytokines studied in the pathogenesis of SLE due to its close link with B lymphocyte maturation into plasma cells and immunoglobulin production. 23 In the lupus-prone model MRL/lpr mice, blockage of IL-33 activity remarkably reduced the IL-1β, IL-6, and IL-17 levels. 12 This study could afford our results. IL-10 and IL-21 have been considered as differentiation factors on B cell differentiation. Several groups found the association of IL-10 and IL-21 serum levels with disease activity scores in SLE.24,42 It is important to note that the role of cytokines in immune response is complex, and several cytokines appear to play paradoxical pathogenic roles in SLE. This may cause their absence of correlation with IL-33.
Previous studies have found that repeated injections of lipopolysaccharide (LPS) in lupus-prone mice could accelerate the development of lupus, increase the production of autoantibodies, and aggravate kidney damage. Immune responses induced by LPS are mediated by TLR4 on target cells. In transgenic mice, TLR4 has been shown to be essential in the production of anti-dsDNA antibodies and in mediating immune complex glomerulonephritis. 16 Enhanced TLR4 signaling alone is considered as a sufficient and a potent trigger to induce SLE.43,44 The latest research reports that IL-33 can promote macrophage maturation and secretion of IL-1β, IL-6, and TNF-α by promoting TLR4 expression. 26 In our study, an apparent positive correlation between IL-33 and TLR4 was also observed. It indicates that IL-33 may involve in the SLE pathogenesis by promoting TLR4 expression.
Our study has several limitations. First, these findings would be strengthened by a larger sample size. Secondly, the expressions of IL-33 and TLR4 were measured at the time of enrollment, and the changes of these proteins before and after treatment were unknown. Thirdly, blocking TLR4 signaling in lupus mice could further identify the proinflammatory pathway of IL-33 in SLE. Therefore, more effort is required on the mechanism of IL-33 and TLR4 in SLE.
Conclusions
We found the level of IL-33 was significantly upregulated in SLE patients. In line with previous studies, there was a positive correlation of IL-33 with inflammatory cytokines IL-4 and IL-6, which act critical roles in the autoantibody production. The cytokines production might be related to the increased expression of TLR4 induced by IL-33. Taken together, upregulated IL-33 expression may contribute to the pathogenesis of SLE by inducing TLR4 expression and promoting the production of inflammatory cytokines.
Footnotes
Acknowledgments
We thank Ying Wang, Qun Su, Yan Huang, Bing Yu, Hongyan Qian, Yuan Liu, Jie Chen, and Guixiu Shi for technical assistance and experimental design. We thank all the volunteers who participated in this study.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Outstanding Innovation Team of Jiangxi Provincial People’s Hospital (No. 19-008) and Natural Science Foundation of Jiangxi Province (20192ACB21006).
Data availability
The data used to support the findings of this study are available from the corresponding author upon request.