
Abstract
The involvement of thromboxane A2 (TXA2) in systemic inflammation and infection is well recognized. However, there are few reports on the involvement of prostanoids in warm shock (the initial pathology of sepsis). Previous studies showed that interleukin (IL)-1β causes a rapid inducible nitric oxide synthase/nitric oxide (iNOS/NO)-mediated relaxation in peripheral blood vessels during warm shock. Furthermore, a transient contraction was seen before this relaxation occurred. The present study aimed to elucidate the mechanism of this transient contraction. We measured isometric tension changes in the superior mesenteric arteries from normal male Wistar rats by adding IL-1β at the point of maximum contraction by phenylephrine (Ph). The same study was performed for each vessel pretreated with various inhibitors, including SQ29548, a TXA2 receptor antagonist, 30 min before Ph contraction. In addition, the concentration of thromboxane B2 (TXB2) in SMA was measured by probe electrospray ionization. Treatment of endothelial vessels with cyclooxygenase 1 (COX1)/2 inhibitors SC560/NS398 and TXA2 receptor antagonist SQ29548 suppressed IL-1β–induced transient contractions. This transient contraction reaction was derived from TXA2. Additionally, gene expression of COX2/TXA2 synthetase and the concentration of TXB2 were significantly increased in IL-1β-exposed vessels. It was demonstrated for the first time in inflamed blood vessels that endothelial cell-derived COX2/TXA2 is induced before iNOS and causes transient contractions. TXA2 may be considered an early sign of warm shock or as a biological defense mechanism in the early stages of septic shock.
Introduction
Despite advances in medicine, sepsis remains the most serious condition encountered in the intensive care unit (ICU), with a mortality rate as high as 40–50%.1,2 Moreover, the incidence of sepsis and septic shock is increasing year by year, making this an important clinical challenge. 3
In the early stages of sepsis, pathogen-associated molecular patterns, such as endotoxin and alarmin, lead to an inflammatory response, which releases inflammatory cytokines around the infected site via various pathways such as those mediated by neutrophils and monocytes. At the site of infection, this is a particularly important biological defense response. However, when systemic inflammation increases and bacteremia develops, the response becomes excessive and harmful. 4 Inflammatory cytokines activate inducible nitric oxide synthase (iNOS) to produce nitric oxide (NO) and cyclooxygenase (COX) to produce prostanoids. These factors relax peripheral blood vessels. 5 The extremities become warm, and the so-called warm shock (hyperdynamic state) progresses.
During warm shock, inflammatory cytokines and excess neutrophil extracellular traps (NETs) damage vascular endothelial cells, and damaged endothelium is shed from the vessel wall. Peripheral arteries lose their normal reactivity due to vascular endothelial cell dysfunction. Cardiomyocytes are also damaged causing reduced cardiac output, lowered blood pressure, and exacerbated peripheral circulatory failure. This is the pathology of cold shock (hypodynamic state). Thus, sepsis interferes with the distribution of systemic blood flow to the organ systems through impaired vasodilation and microcirculation. 6
Warm shock does not last long and shifts to cold shock within a few hours. Therefore, early recognition of sepsis is important, and treatment is required early to maintain homeostasis. However, there are no effective therapeutic drugs specific to sepsis or markers that can accurately predict its severity. Thus, an appropriate early diagnosis of sepsis cannot be made. To solve this problem, elucidation of sepsis pathophysiology is both necessary and urgent.
In previous studies, we showed that IL-1β causes a rapid iNOS/NO-mediated relaxation of the superior mesenteric artery (SMA) during warm shock. 7 Interestingly, a transient contraction was observed before this relaxation in rat SMAs, but the mechanism of this contraction remained unclear.
Here, we focused on thromboxane A2 (TXA2), an inflammatory mediator with vasoconstrictor activity. TXA2 is an arachidonic acid (AA) metabolite under the prostanoids subclass involved in various immune and inflammatory reactions. When platelets and vascular endothelial cells are stimulated by inflammatory cytokines, phospholipase A2 produces AA from cell membrane phospholipids. AA is converted to prostaglandin H2 (PGH2) by activation of COX and further converted from PGH2 to TXA2 by thromboxane synthase. The released TXA2 binds to the thromboxane receptor (TP) on the platelet surface and induces platelet aggregation by a G protein-mediated signal.8,9 Although TXA2 is mostly synthesized in platelets, TXA2 and TP receptor expression is also found in vascular endothelial and smooth muscle cells.10,11 TXA2 is known to have a strong vasoconstrictor effect in addition to its platelet aggregation effect.12,13 Thus, the involvement of TXA2 in systemic inflammatory conditions, such as infectious diseases, is well recognized. However, there are few reports on the involvement of prostanoids in warm shock. In the present study, we hypothesized that this transient contraction was derived from TXA2 and might be an early sign of warm shock. The purpose of present study was to elucidate the mechanism of transient contraction by assessing the temporal changes in the contraction–relaxation response of inflamed blood vessels.
Materials and methods
Sample size calculation and justification
The sample size was established using power analysis; variance explained by special effect and variance within groups were used to measure sample variability; type 2 error at p < 0.05; one-way ANOVA with 90% analysis capacity was used. The sample size was calculated using the G* Power Version 3.1.9.6 software.
Animal procedures
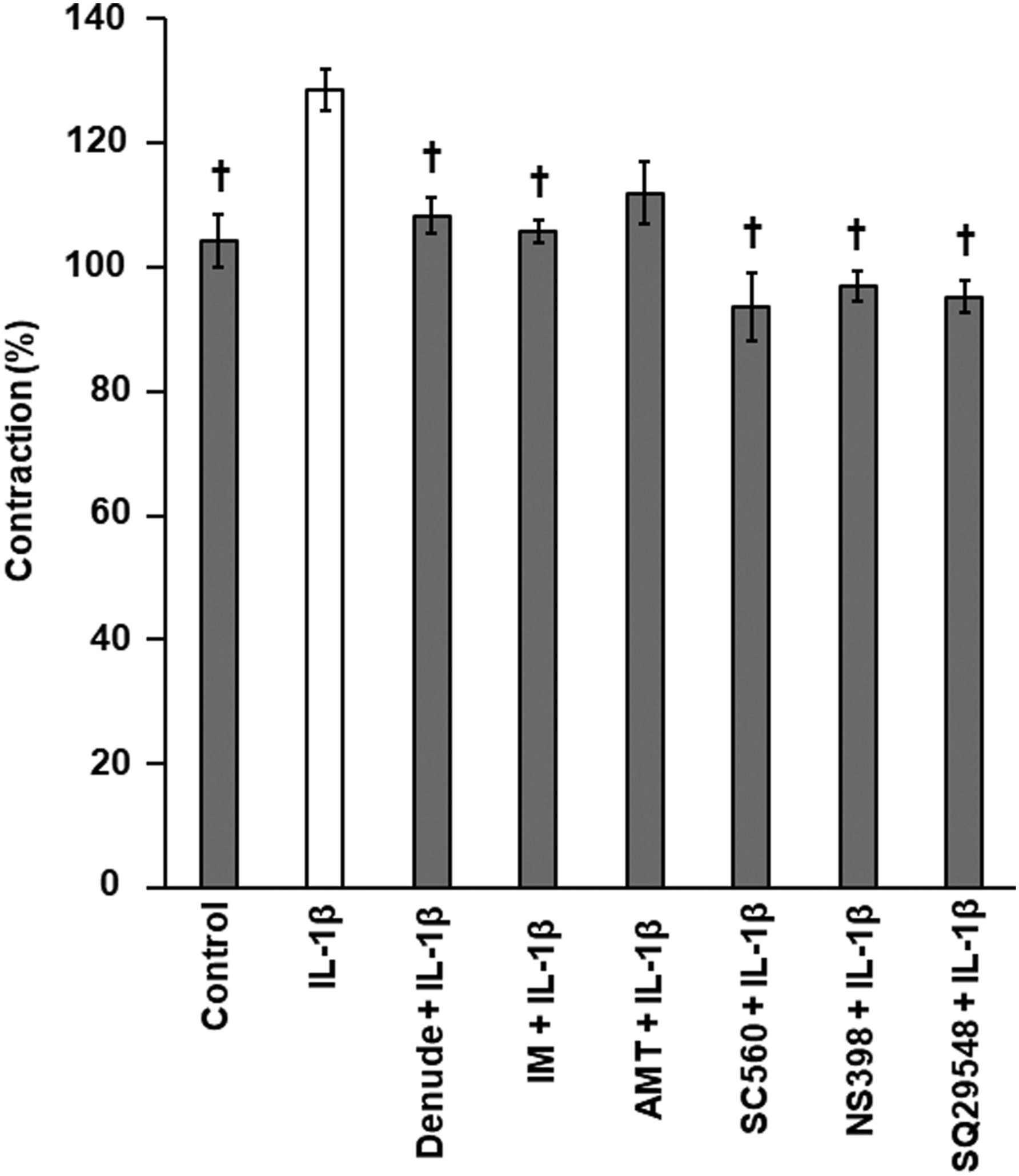
All protocols for the animal experiments were approved by the Animal Care Committee of Nara Medical University in accordance with the policies established in the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (Permit No. 12859). Male Wistar rats (10–12 weeks old, 330–350 g, total number of rats n = 48; Figure 1 and Figure 2 (n = 8), Figure 3 (n = 8), Figure 4 (n = 8), Figure 5 (n = 24); CLEA Japan, Inc., Tokyo, Japan) were placed in a quiet, temperature- and humidity-controlled room (24°C and 58%, respectively) maintained under a 12–12 h light–dark cycle (light between 08:00–20:00). In total, 48 rats were utilized since each data set used in a single analysis had to be obtained from 4–8 samples. Representative traces showing the effect of time-dependent changes in IL-1β–induced vasoconstriction. (a) The trace showing the continuous vascular reactivity of SMA from exposure to IL-1β until 3 h after. (b) Box-and-whisker plot showing the effects of various inhibitors and endothelium denudation on IL-1β–induced vasoconstriction from Panel 1(a) using four time points: 0, 1, 2, and 3 h. *p < 0.05 vs Control. †p < 0.05 vs IL-1β. n = 8 (rings). SMA: superior mesenteric arteries; Ph: phenylephrine; IL: interleukin; IM: indomethacin; SQ29548: Thromboxane A2 receptor antagonist. Summary data showing the effects of various inhibitors and endothelium denudation on IL-1β–induced transient contractions. Results represent the mean +SEM and are expressed as a percent of the peak contraction elicited by 10−5 M Ph. †p < 0.05 vs IL-1β. n = 8 (rings). SEM: standard error of mean; Ph: phenylephrine; IL: interleukin; IM: indomethacin; AMT: inducible nitric oxide synthase inhibitor; NS398: cyclooxygenase 1 inhibitor; SC560: cyclooxygenase 2 inhibitor; SQ29548: Thromboxane A2 receptor antagonist. Effect of IL-1β on the corresponding concentration-response curves to L-arginine. Results represent the mean +SEM *p < 0.05 vs Control. n = 9 (rings). SEM: standard error of mean; IL: interleukin; Chx: cycloheximide. Relative mRNA expression profiles of COX1, COX2, TBXAS, PTX3, SOD1, and SOD2 in SMAs. (a) Expression levels were determined by real-time PCR and then normalized to the expression of Hprt1. For quantification of COX1, COX2, and TBXAS expression, the 2−ΔΔCt method was used. Data were expressed relative to the value for SMA not exposed to IL-1β. Results represent the mean +SEM *p < 0.05 vs 0 h. n = 4 (rats). (b) Expression levels were determined by real-time PCR and then normalized to the expression of Hprt1. For quantification of PTX3, SOD1, and SOD2 expression, the 2−ΔΔCt method was used. Data were expressed relative to the value for SMA not exposed to IL-1β. Results represent the mean +SEM *p < 0.05 vs Control. n = 4 (rats). mRNA: messenger RNA; SEM: standard error of mean; COX1: cyclooxygenase 1; COX2: cyclooxygenase 2; TBXAS 1: thromboxane A synthase 1; PTX3: pentraxin 3; SOD1: Cu/Zn superoxide dismutase; SOD2: Mn superoxide dismutase; SMAs: superior mesenteric arteries; PCR: polymerase chain reaction; HPRT: hypoxanthine phosphoribosyl transferase; IL: interleukin; Ct: comparative cycle threshold. Determination of TXB2 levels in SMAs. TXB2 was measured using a PESI-MS/MS. *p < 0.05 vs Control. n = 8 (rats). SEM: standard error of the mean; IL: interleukin; IL: interleukin; IM: indomethacin; SMAs: superior mesenteric arteries; TBX2: thromboxane B2.



Isolated superior mesenteric artery ring preparation
The rats were euthanized by exsanguination. SMA was excised, and its adherent connective tissues were removed. Each SMA was separated from the surrounding connective tissue and fat, cut into 8–9 rings, and then mounted on an organ bath system. Subsequently, the vascular function was assessed by measuring the vascular isometric force, as reported previously.14,15 SMA was sectioned into rings 1–1.5 mm long. The rings were horizontally mounted on 50-μm-diameter tension hooks in 4-mL organ baths containing Krebs–Ringer solution (118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 25 mM NaHCO3, 2.5 mM CaCl2, and 10 mM D-glucose; pH 7.4). The solution was maintained at 37°C by a thermally regulated water circuit and continuously aerated with 95% O2 and 5% CO2.
SMAs were used in this study to determine the isometric tension. This major artery of the abdomen was chosen because there is considerable cumulative knowledge of its vascular function. Isometric tension was monitored with a force-displacement transducer (Primetech Co., Tokyo, Japan) to which one side of each tension hook was connected, and documented with a pen recorder (Nihon Kohden Kohgyo Co., Tokyo, Japan). SMA rings were suspended by the hooks, the tension was set to 0.2 g, and the rings were stabilized in the organ baths at 37°C for 90 min. The bath fluid was changed every 15 min. The resting tension was maintained at 0.2 g throughout the experiment. 15 To examine the role of the endothelium in IL-1β–induced contraction and relaxation, it was denuded by gently rubbing the intimal surface with a stainless-steel wire. Removal of the endothelium was confirmed by the loss of relaxation in response to 10–6 M acetylcholine. For the studies on endothelium-intact vessels, the rings were discarded if acetylcholine-induced relaxation was not ≥80%. No SMA rings were repeatedly measured.
Tension measurement
We previously reported that IL-1β exposure has an attenuating effect on phenylephrine (Ph)-induced contractions. 16 To eliminate this effect in this study, IL-1β was added when the plateau tension was reached after pre-contraction with Ph. Using segments where the presence or absence of endothelial cells was confirmed, time-course observations were performed after IL-1β exposure. Arteries were pre-contracted with 10−5 M Ph. At peak pre-contraction, 20 ng/mL of IL-1β was added to the organ baths. Thereafter, tissue tension was monitored for 3 h. Various inhibitor combinations were tested using different SMA rings isolated from the same rat. Inhibitors were added to the bath 30 min before Ph. Concentrations are reported as the final molar concentration in the organ bath. The transient contractions were expressed as a % of the peak contraction in response to 10−5 M Ph. All drugs were co-incubated with IL-1β over the 3 h period.
In addition, L-arginine was added cumulatively after 3 h. Dose-response curves for L-arginine were also constructed. The relaxation was expressed as the rate of contraction before L-arginine was added.
Quantitative real-time polymerase chain reaction
Following SMA removal (n = 8 rats), the segments, divided into three, were incubated in Krebs–Ringer’s solution containing 20 ng/mL of IL-1β for 0.5–3 h at 37°C via a thermally regulated water circuit and continuously aerated with 95% O2 and 5% CO2. Total RNA was extracted from SMA samples with the RNeasy® Mini kit (Qiagen, Valencia, CA, USA). Total RNA (5 ng) was reverse-transcribed to cDNA with a SuperScript™ RT kit (Life Technologies, Carlsbad, CA, USA) using random hexamers as described in the manufacturer’s protocol. To measure COX1, COX2, thromboxane-A synthase (TBXAS), pentraxin 3 (PTX3), superoxide dismutase 1 (SOD1), and SOD2 expression, qRT-PCR was performed in a StepOne™ system (Applied Biosystems, Foster City, CA, USA) using TaqMan® probes and the primers TXBAS (Tbxas 1; assay ID = Rn00562160_m1), PTX3 (Ptx3; assay ID = Rn01769850_m1), COX1 (Ptgs1 assay ID = Rn00566881_m1), COX2 (Ptgs2 assay ID = Rn01483828_m1), SOD1 (Sod1; assay ID = Rn00566938_m1), and SOD2 (Sod2; assay ID = Rn00690588_g1). Hypoxanthine phosphoribosyl transferase (Hprt1; assay ID = Rn01527840_m1) was the endogenous control. To quantify TXBAS, PTX3, COX1, COX2, SOD1, and SOD2 expression, the 2−ΔΔCt method was used. 17
Determination of thromboxane B2 levels in superior mesenteric artery
Following SMA removal (n = 24 rats), the segments, divided into three, were incubated in Krebs–Ringer’s solution containing 20 ng/mL of IL-1β for 1–3 h at 37°C via a thermally regulated water circuit and continuously aerated with 95% O2 and 5% CO2. Each segment was homogenized and centrifuged (17,900 g, 20 min, 4°C), and the supernatant was used for analysis. A Shimadzu 8045 triple quadrupole mass spectrometer with a Probe electrospray ionization (PESI) ion-source (Shimadzu, Kyoto, Japan) was used for direct TXB2 detection. The samples were placed directly on a dedicated plastic sample plate and set on the PESI ion-source. 18 The probe needle was lowered such that the needle tip touched the sample and then raised to apply a high voltage for ionization. This vertical movement was repeated, and the generated ions were introduced into the tandem mass spectrometry (MS/MS) system. The PESI-MS/MS conditions were set as follows: Probe-applied voltage, 3.0 kV; cycle time for probe movement, 160 ms; desolvation line temperature, 250°C; heat block temperature, 50°C; and polarity, negative. The probe position (distance from the tip of the needle to the center of the MS inlet) was set at y-axis 2 mm and x-axis 2.5 mm. The product ion (Pl) spectra of the compounds were measured in the multiple reaction monitoring (MRM)-Pl scan mode. Collision energies were adjusted to optimize the product ion signal as −18 eV for TXB2. The MRM mode was used to monitor the transition of the deprotonated molecule at m/z 369.2 → 169.1. 19 The peak areas were calculated by constructing a calibration curve using standard controls and then by integrating all peaks for each compound per run using the built-in LabSolutions software (ver. 5.99 SP2, Shimadzu Corp., Kyoto, Japan). The probe needle was changed between samples to prevent contamination. Protein contents were measured with the Qubit Protein Assay Kit (Invitrogen, Waltham, USA).
Statistical analysis
Results are expressed as mean ± standard error of the mean (S.E.M.). Statistical analyses were performed using the Student’s t-test and one-way analysis of variance (ANOVA). Post-hoc comparisons were performed using Bonferroni’s multiple comparison test. The results were considered statistically significant at a p value below 0.05.
Chemicals and drugs
Drug application
IL-1β, Ph, and ethanol were obtained from Wako Pure Chemical (Osaka, Japan). Cycloheximide, indomethacin (IM), nitro-L-arginine, SQ29548, NS398, SC560, and 2-amino-5,6-dihydro-6-methyl-4H-1,3-thiazine hydrochloride (AMT) were obtained from Sigma-Aldrich (St Louis, MO, USA). TXB2 was obtained from Cayman Chemical (No. 19030, Ann Arbor, MI, USA).
Results
We observed the continuous vascular reactivity of SMA after exposure to IL-1 β for 3 h (Figure 1(a)). In addition, exposure to IL-1β in Ph-treated pre-contracted SMAs induced transient contractions after 1 h. After that, it recovered to the same level of tension as the Ph pre-contracted SMAs. Figure 1(b) represents the changes in tension in Figure 1(a) as four time points at 0, 1, 2, and 3 h. Ph-induced pre-contraction SMAs showed significant transient contraction responses 1 h after IL-1β exposure, and treatment with the nonselective COX inhibitor IM (10−5 M) and the TP antagonist SQ29548 (10−6 M) suppressed transient contractions. However, this transient contraction reverted to the same contractions seen at the 0 h time point before IL-1β exposure at about 2 h after IL-1β exposure (Figure 1(b)). Vascular reactivity showed a dramatic relaxation response at 3 h after IL-1β exposure. Furthermore, vascular reactivity showed a significant relaxation response at 3 h in both IL-1β and IM-treated SMAs. This relaxation was consistent with our previous reports. In addition, the relaxation response was significantly suppressed by SQ29548 treatment. This suggests that TP, which is a receptor for TXA2, was involved not only in the transient contraction that occurred at 1 h due to IL-1β exposure but also in the relaxation at 3 h.
Figure 2 shows the effect of various inhibitor treatments on the transient contractions at 1 h, as shown in Figure 1(b). Treatment of denuded endothelium, IM, COX1/2 inhibitors NS398 (10−5 M)/SC560 (10−6 M), and TP antagonist SQ29548 suppressed transient contractions (Figure 2). Transient contractions were not suppressed by the iNOS inhibitor AMT (5 × 10−5 M).
The relaxation of NO was examined to confirm whether IL-1β exposure affected the relaxation response after 3 h (Figure 3). L-Arginine (a substrate of NO)-induced relaxation was investigated. Vascular reactivity showed a significant relaxation compared to control at 3 h after IL-1β exposure. The relaxation induced by IL-1β disappeared with endothelium denudation and was suppressed by cycloheximide (10−4 M), which is a protein synthesis inhibitor and was not different from that in the control (Figure 3).
Quantitative real-time polymerase chain reaction analysis revealed that COX1 mRNA expression was slightly increased in SMA treated with IL-1β for 1 h compared to untreated SMA, but not significantly. However, COX2 mRNA expression was significantly increased in SMA treated with IL-1β for 1 h. Expression of TBXAS1 mRNA, which is downstream of the prostanoid production pathway and is a TXA2 synthase, was significantly increased in SMA treated with IL-1β for 1 h (Figure 4(a)). In addition, we investigated the expression of SOD1, SOD2, and PTX3, which are markers of the effects of inflammation caused by IL-1β exposure. Expression of SOD2 and PTX3 mRNA was significantly increased in SMA treated with IL-1β for 1 h (Figure 4(b)).
Due to the short half-life and quantification difficulty of TXA2 protein, a mass spectrometer was used to measure the protein concentration of the more stable TXB2 produced via TXA2 hydrolysis. Our results showed that the concentration of TXB2 was significantly increased by IL-1β treatment compared with that in the control. In addition, the IM-treated SMAs reduced the TXB2 concentration, which was not different from that in the control (Figure 5).
Discussion
In this study, we demonstrated that endothelial cell-derived TXA2 induces transient vasoconstriction under Ph-mediated contraction in inflamed blood vessels. This phenomenon is a precursor to a dramatic relaxation that occurs later.
mRNA levels of PTX3 and SOD2 increased approximately 1 h after IL-1β exposure, but no change in endothelial NOS (eNOS) expression was observed, indicating that we successfully recorded the temporal changes in the early stages of the inflammatory vascular response of unimpaired endothelial cells.
Transient contractions in SMA were suppressed via endothelium removal and TXA2-related inhibitors. In addition, 1 h post-IL-1β exposure, the mRNA levels of COX2 and TXA2 synthetase in SMA were increased. Increased levels of TXB2 protein, a TXA2 metabolite, in SMA persisted for 1–3 h following IL-1β exposure. Therefore, this transient contraction might have been caused by the induction of COX2 by IL-1β stimulation and the subsequent increase in TXA2 production in endothelial cells. These results are consistent with reports of early increased production and release of COX2, TXA2, and TXB2 in the lungs and arteries during lipopolysaccharide (LPS)-induced endotoxic shock.20–22
Other reports have described that the expression of COX2 and iNOS is associated with several pathologies that occur in sepsis. 23 Furthermore, these have been shown to underlie the relaxation of cerebral arteries after LPS exposure.22,24 Similarly, our previous report showed that the dramatic relaxation after 3 h is an iNOS/NO-mediated reaction. 7 Vo et al. 25 showed that LPS caused NO-mediated relaxation of the rat aorta. However, no transient contraction was observed in the aorta. Based on these results, we speculated that it is difficult to detect minute changes in blood vessels during warm shock in the aorta, which is the conduit artery. Therefore, for the first time, we were able to capture transient contractions in peripheral blood vessels. Furthermore, we found a change in the contraction–relaxation of inflammatory vascular response at four time points.
It is unclear whether transient contraction by TXA2 is completely independent of the dramatic relaxation reaction 3 h after IL-1β exposure or if it is associated with relaxation. Since the dramatic relaxation reaction occurs 3 h after IL-1β exposure and almost disappears in the presence of a TP antagonist, the former theory is unlikely, and the transient contraction is considered to be associated with the subsequent relaxation. This is thought to occur as inflammation causes an increase in endothelial cell-derived TXA2, which in turn causes transient contraction through the activation of the TP in the vascular smooth muscle. Furthermore, TP activation is considered to activate iNOS, which produces a large amount of NO in the smooth muscles and increases relaxation. 25,26,27 This is consistent with reports that COX2 is induced more rapidly than iNOS and that early production of prostanoids stimulates iNOS expression. 28 It has been reported that in LPS exposure, the contractile prostanoid TXA2 is induced by COX2 to compensate for the decrease in contraction due to the increase in NO content from iNOS expression. 29 However, in this study, we used time-course experiments to show for the first time that in inflammatory arteries, exposure to IL-1β after Ph-pre-contraction induces endothelial cell-derived COX2/TXA2 before iNOS. We also demonstrated that COX2/TXA2 causes transient contractions and regulates subsequent iNOS/NO-mediated relaxation. A limitation of this study was not investigating the mechanism that causes the iNOS/NO-mediated relaxation reaction by TXA2 in detail. However, other groups have previously reported the activation of eNOS by TXA2 in endothelial cells. 30 It has also been reported that the activity of eNOS in endothelial cells produces excess iNOS/NO through the activity of the inflammatory transcription factor nuclear factor-κB, which relaxes blood vessels and induces shock. 26 In this study as well, it is suggested that iNOS/NO level may have increased after eNOS was activated by TXA2 contraction.
Blood vessels maintain vascular tonus by constant competition between contraction and relaxation. Contraction and relaxation always offset each other, and the final state of vascular tonus appears as blood pressure. In addition, peripheral vascular contraction and relaxation are also associated with changes in the microcoronary arteries. The peripheral vascular reactivity shown in this study is thought to reflect systemic hemodynamics in the early stages of septic shock. Therefore, the findings of this study may have potential ramifications in other disease entities such as cardiovascular disease. To elucidate the hemodynamics during warm shock, it is necessary to examine the effects of other factors associated with sepsis, 31 such as reactive oxygen species and histones, major components of NETs.
In addition, it was difficult to evaluate the expression of factors such as TXA2, which is a contractile protein, under transient contraction. Therefore, we evaluated related factors through gene expression analysis and mass spectrometry and found that the transient contraction was a TXA2-mediated response. Furthermore, it is possible that the transient contraction did not occur only due to TXA2 but was a synergistic effect of TXA2 and other factors. In recent years, a novel endothelium-derived vasoactive factor called uridine adenosine tetraphosphate (Up4A) that activates purinergic receptors has been identified. Up4A has been shown to cause contraction via the receptors P2X and P2Y.32,33,34 Furthermore, TXA2 has been shown to activate TP through activation of the receptors P2X and P2Y. 35 Therefore, it is speculated that Up4A is associated with TXA2 and causes this transient contraction. The effect of Up4A on transient contraction has not been ruled out. Moreover, it is necessary to evaluate the expression of Up4A and suppression of transient contraction following treatment with an antagonist of receptor P2.
The lack of in-depth research of transient SMA contraction by TXA2 in rat models of sepsis is a limitation of this study. It is necessary to investigate the expression levels of SMA TXB2 protein in sepsis models.
Conclusions
In conclusion, it was shown that in inflamed blood vessels under Ph-induced contraction, TXA2/COX2 from the endothelial cells induces transient vasoconstriction after IL-1β exposure. It is suggested that TXA2 may be an early sign of warm shock or as a biological defense mechanism in the early stages of septic shock. In fact, despite substantial progress in the understanding of the pathophysiology of sepsis, no unique biomarker is available for diagnostic needs. In this study, we found that this transient contraction is derived from TXA2 and provides a guide for the treatment of warm shock. The results of the study indicate that TXA2-induced transient contraction in the endothelial cells of inflamed vessels may offer new insights into vascular function regulation. However, we have not investigated the crosstalk between TXA2 and other factors such as Up4A in the early stages of septic shock. Conversely, we were able to observe the changes over time in the contraction of inflammatory blood vessels, which will help better our understanding of the pathophysiology of sepsis.
Footnotes
Acknowledgements
Author contributions
KY, RK, and SK designed the study, developed the methodology, performed the analysis, and wrote the article. KY and RK collected the data.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Japan Society for the Promotion of Science KAKENHI (Grant No. 18K17356 to KY).
Ethical approval
Ethical approval for this study was obtained from the Animal Care Committee of Nara Medical University (Approval Number/12859).
Animal welfare
The present study followed international, national, and/or institutional guidelines for humane animal treatment and complied with relevant legislation.