
Abstract
A lithium metal anode has a high energy density, but its applications face numerous challenges, particularly the irregular deposition of lithium dendrites, which form due to the aggregation of lithium clusters, posing serious safety risks. Research has revealed that small lithium clusters have the potential to enhance the electrode potential in Lithium-ion batteries (LIBs), and cluster energy affects the stability of lithium clusters. Modern computational tools and DFT calculations of lithium clusters can provide invaluable insights into their chemical, physical, and structural properties. However, calculating structural properties through large-scale experiments can be time-consuming and expensive. To overcome the challenges inherent in studying lithium clusters, this study employs a novel approach. We construct molecular graphs for the most stable structures and leverage CoM Polynomial computations to generate codescriptors, enabling efficient and accurate predictions of cluster energy for diverse lithium cluster configurations. The curvilinear regression analysis filters out highly significant regression equations and highly predictive codescriptors. Additionally, we investigated two molecular structures of porous graphene, namely linear and triangular, and obtained topological codescriptors’ analytical expressions by utilizing the CoM Polynomial in each case. A deeper understanding of both lithium clusters and porous graphene properties is essential for optimizing LIBs’ performance and stability.
Introduction
In recent years, the remarkable physical and chemical properties of clusters have sparked significant interest, leading to rapid developments in interdisciplinary research.1,2 Clusters act as an intermediate transition state in the transformation of substances from atoms and molecules to macroscopic objects, representing a novel material structure with diverse applications in fields like chemical sensing, optical and optoelectronics, fluorescence, biomedical, catalysis, solar cells, energy, and environment.3–9 Of particular interest are small metal clusters, especially lithium clusters, which can form with various elements and appear in processes like electrodeposition and condensation. For instance, lithium clusters form on the anode of a lithium battery during charging, but they can also cause stability issues, such as the growth of lithium dendrites.10,11 Earlier studies have shown that lithium cluster properties, like binding energy, cluster energy, and stability, are related to their size, with most research focusing on small clusters due to computational complexity limitations.12,13 Density functional theory and other calculation methods have been used to study various properties of lithium clusters, including ground and excited state geometry, electronic structure, binding and dissociation energies, ionization potentials, total energy, and cluster energy for different cluster sizes.14–19 However, these techniques are computationally expensive and time-consuming.
Research into better electrode materials for batteries has led to the discovery of carbonaceous nanomaterials. Among these, graphene stands out for its exceptional properties, including high specific surface area, excellent electrical conductivity, mechanical strength, and chemical stability.20–23 However, graphene’s tendency to stack together inhibits its electrochemical performance. To overcome this drawback, researchers have explored porous graphene materials, which enable electrolytes to penetrate electrodes and interact with active materials efficiently, facilitating electron transfer and ion diffusion. 24 In 2009, Bieri et al. 25 created a porous graphene structure (Figure 1) by altering the graphene structure, and its electronic properties were subsequently studied in Du et al., 26 leading to nano electronic applications being discovered.

(a) Structure of a fraction of the porous graphene network and (b) STM image of porous graphene.
Graphene-based porous materials possess the advantages of both graphene and porous materials, have been designed and developed with high porosity over time to offer better performance, and have unique properties that enable them to be used for a variety of applications such as its high surface area facilitates efficient adsorption, enabling applications in water purification, 27 catalysis, 28 and sensing. 29 Tunable pore sizes enable selective molecule capture, which is particularly important in gas separation. 30 Additionally, its excellent electrical conductivity supports electron transfer in super capacitors. 31 Porous graphene is discussed in detail in Huang et al. 32 and Zhang et al. 33
Lithium cluster properties are crucial for advancing battery technologies, and understanding their structural behavior also provides valuable insight into fundamental areas such as cluster formation and stability and nanomaterials design. Similarly the wide range of applications of porous graphene structures, with significant potential for advancements, necessitates studying their properties. Using quantitative structure-property relationships (QSPR) as a computational tool, researchers can predict nanostructure properties solely based on their chemical structure and molecular descriptors, enabling them to design nanostructures that have specific properties without having to actually synthesize them. 34 Chemical graph theory employs molecular graphs to represent chemical structures, with vertices symbolizing atoms and edges as bonds and topological molecular descriptors transform these graphs into mathematical real numbers, facilitating the prediction of properties for chemical structures.35,36 Wiener’s 37 pioneering work introduced the Wiener index, the first topological molecular descriptor, enabling the prediction of boiling points for alkenes. This marked a significant leap in understanding the relationship between molecular structure and properties. Since then, numerous individual topological descriptors have been developed. 38 Notably, in 2015, Deutsch and Klavžar 39 developed the M-Polynomial technique, a powerful tool that provides numerous degree-based topological descriptors at once. Building upon the success of the M-Polynomial technique, the field of chemical graph theory has recently witnessed the introduction of two exciting new tools: the NM-Polynomial 40 and the CoM Polynomial. 41
In Shanmukha et al.,42,43 the computation of M Polynomial and NM Polynomial for porous graphene was conducted. Subsequently, in Govardhan and Roy, 44 adjustments were made to the edge partition by calculating the entropy of porous graphene. Some other graph property-based topological descriptor were computed for polyphenylene honeycomb structure in Krishnan and Rajan, 45 Krishnan et al., 46 and Aftab, 47 and for porous nanographenes in Balasubramanian. 48 The structural and electronic properties of Lithium clusters up to ten were computed using density functional theory in Chetri et al., 49 while the M-Polynomial computation of these clusters can be found in Chetri et al. 50
This article presents a twofold objective. Firstly, we propose a novel, computationally efficient, and alternate method for calculating the cluster energy of Lithium clusters. Secondly, we aim to address the gap in current literature by computing the CoM Polynomial of various porous graphene structures. This combined investigation is motivated by the significant role of both Lithium clusters and porous graphene in energy storage devices. Section “Preliminaries” lays the groundwork in graph theory, providing the necessary background for understanding the results presented in subsequent sections. Section “Relationship between the cluster energy with various topological codescriptor of lithium clusters” explores the relationship between specific Lithium cluster properties and the topological codescriptors obtained from the CoM Polynomial. Finally, Section “CoM polynomial of 2D porous graphene” presents detailed analytical results for linear and triangular porous graphene structures. This section includes both the computation of the CoM Polynomial and numerical comparisons in each case.
This article aims to develop a new method for calculating the cluster energy of lithium clusters and compute the CoM Polynomial for porous graphene structures. The motivation comes from the importance of lithium clusters and porous graphene in energy storage devices. The article starts with a background in graph theory, then explores the relationship between lithium cluster properties and topological codescriptors from the CoM Polynomial. Finally, it presents analytical results for linear and triangular porous graphene structures, including the computation of the CoM Polynomial and numerical comparisons (Figure 2). This work combines quantum chemistry and graph theory to advance battery technology.

Overview of method used in this manuscript.
Preliminaries
In this article,

where



and from Berhe and Wang
51

From (1) several degree-based topological codescriptor (TCD) as shown in Table 1 can be obtained
List of topological codescriptor with notation obtained from CoM polynomial.

In Section “Relationship between the cluster energy with various topological codescriptor of lithium clusters” and “CoM polynomial of 2D porous graphene” the molecular graph of Lithium cluster is denoted as G
Relationship between the cluster energy with various topological codescriptor of lithium clusters
Molecular descriptors are crucial for predicting material features and designing economically, enabling rapid material discovery, saving time and costs compared to other techniques. This approach is particularly valuable in early material design stages, helping researchers focus on promising candidates and allowing to optimize conditions to enhance the likelihood of obtaining materials with desired properties. This section emphasizes an investigation conducted to assess the predictive capacity of codescriptors derived from CoM Polynomial. The inquiry aims to ascertain whether these codescriptors hold significance in predicting properties and whether they merit utilization in various applications. To achieve this we have constructed the molecular graph from the most stable structures of Lithium clusters49,52 as shown below in Figure 3.

Molecular graph of
The CoM Polynomial of the molecular graphs (Figure 3) was computed directly by employing the edge partition method. For example, the CoM Polynomial for the molecular graph of Lithium cluster
This section investigates the predictive power of CoM Polynomial-derived codescriptors for material properties. Molecular graphs of stable Lithium clusters were constructed and their CoM Polynomials computed. The approach aims to determine if these codescriptors can predict material properties, enabling rapid material discovery and optimization.
The edge set of

Using 2, we have
By the definition of CoM Polynomial

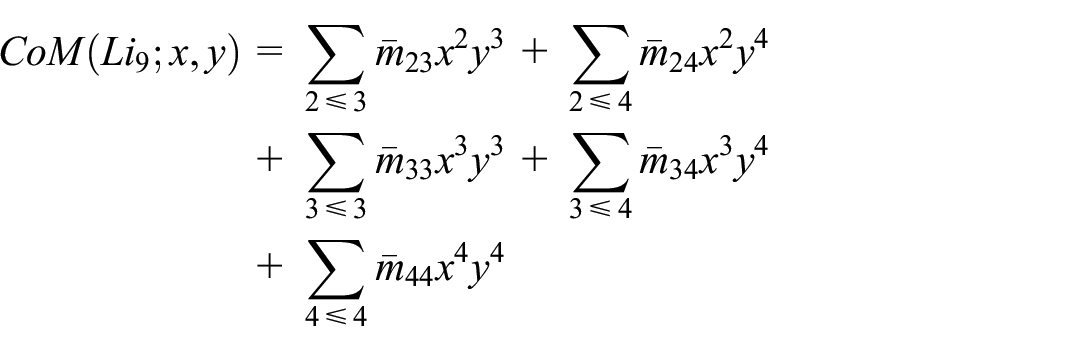
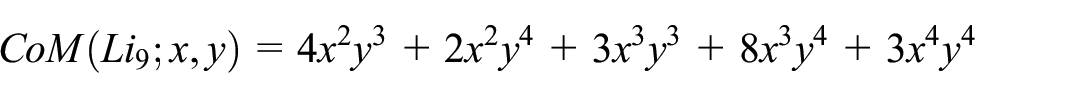
Below Table 2 lists ten codescriptor such as

then
Lithium clusters

By the use of Table 1, we get
The cluster energy (CE) of a Lithium cluster required to assemble a specific number of lithium atoms into a cluster. Table 2 listed the values of CE of

Equation (3) is the general form of the regression model, where
The findings in Tables 3 and 4 indicate that the Symmetric Division codescriptor
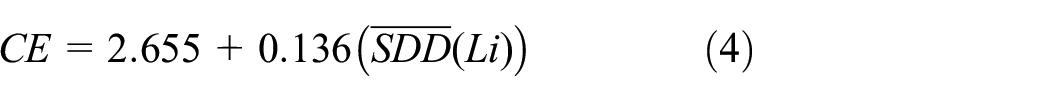


Square of correlation coefficient

Statistically predicted regression models that give best estimate for cluster energy.

Figure 4 shows plots of the linear, quadratic, and cubic regression equations (4), (5), and (6) of the Cluster energy with

Plot of: (a) linear, (b) quadratic, and (c) cubic regression equations for the cluster energy of lithium clusters predicted by best codescriptor.
Equations (4), (5), and (6) can be used to calculate the CE of Lithium clusters by using codescriptor such as
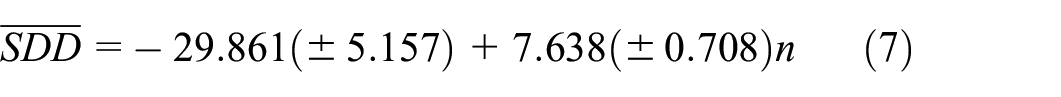


Figure 5 shows plots of the linear, quadratic and cubic regression equations (7), (8) and (9) between
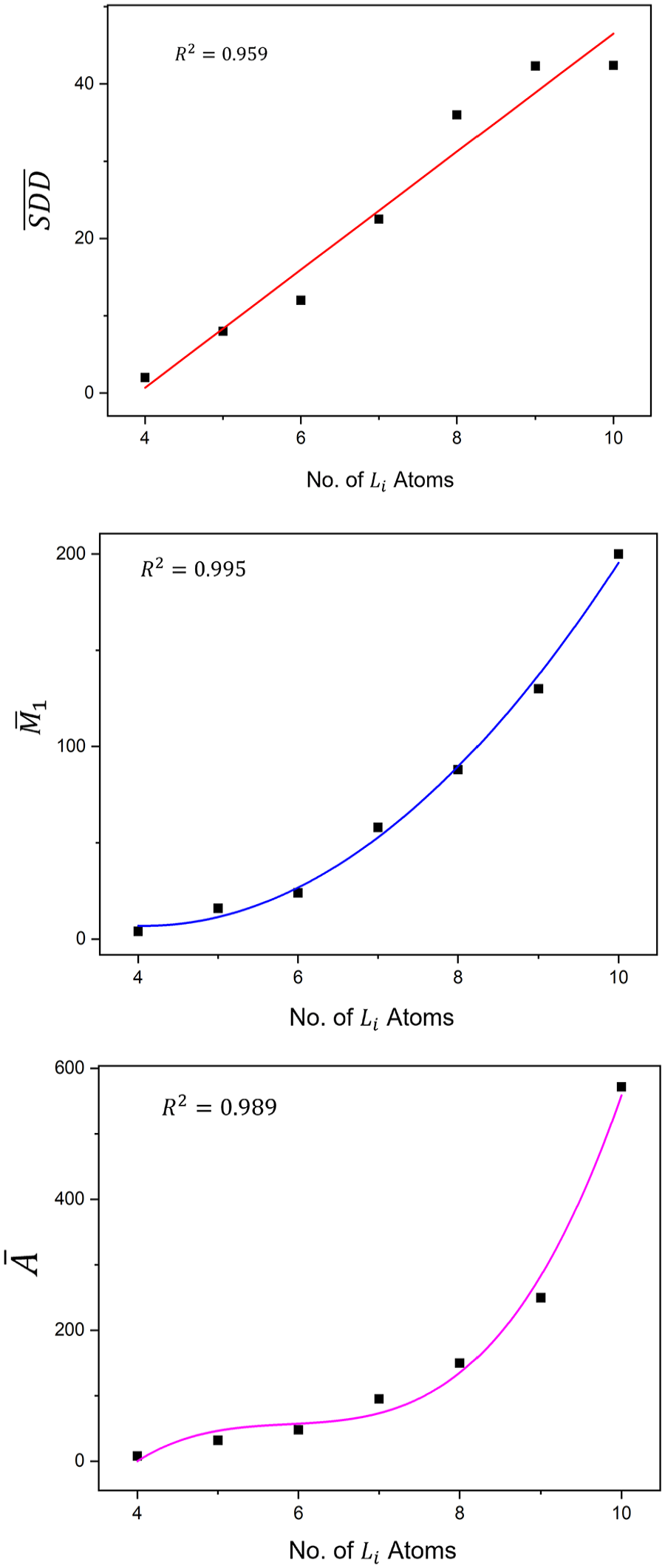
Plots of the linear, quadratic, and cubic regression equations (7), (8), and (9) between
Using equations (7), (8), and (9) for any number of Li atoms in a Lithium clusters one can find the values of
CoM polynomial of 2D porous graphene
In this section, we develop analytical expressions for topological codescriptors (TCD) derived from the CoM Polynomial, for various porous graphene structures. Subsequently, we numerically tabulate TCDs for these structures, followed by graphical representation and analysis to identify trends across different structures. A porous graphene is a material with nanopores in the plane, and the pore size and distribution of the porous graphene are determined by the method of production. Bieri et al. 25 successfully transformed graphene structure into a porous graphene structure by strategically removing phenyl rings at regular intervals. The resulting structural relationship between this network (represented by bold lines) and the original graphene (depicted with thin lines) is visually illustrated in Figure 6.

Structure of porous graphene.
The unique properties of porous graphene, applicable across diverse fields, have recently sparked a surge in scientific interest. This has driven researchers to develop efficient methods for synthesizing well-defined porous graphene with controlled pore structures. By adopting various approaches, researchers have succeeded in generating diverse types of porous graphene structures such as ordered porous graphene nanoribbon 53 and trigonal porous graphene 54 as evidenced by the literature. 55 These structures inspired us to conduct a topological study using CoM Polynomial techniques. The structures examined in this section are depicted in Figure 7.

Structure of linear and trigonal porous graphene.
CoM polynomial of linear porous graphene (LPG)
As a first step, we determine the expression for the CoM polynomial for LPG.
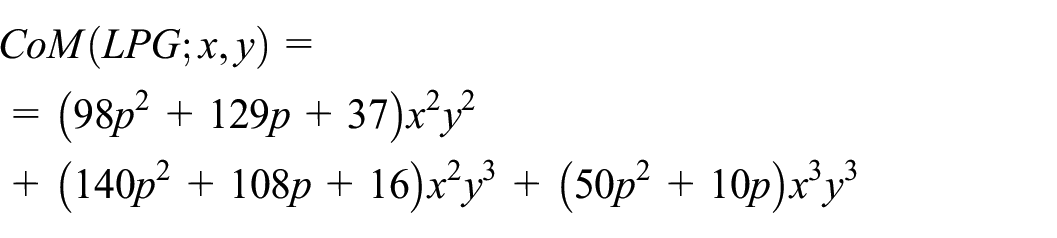



Molecular graphs G
In a similar way, LPG’s vertex set can be divided into two classes according to their degrees.

Using (1), we have

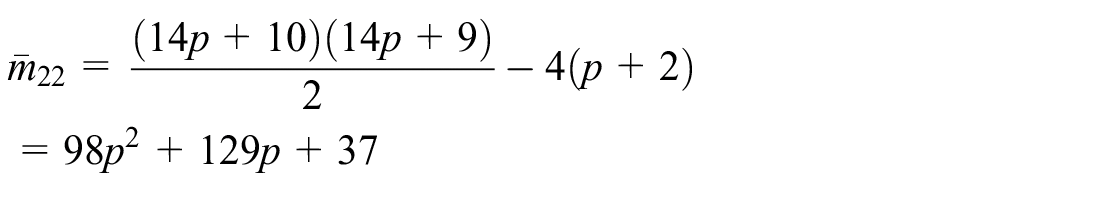
Similarly,
By definition of Co-M-polynomial


Thus, the result.
The next proposition uses Theorem 1 to recover some degree-based TCD of the LPG.

then
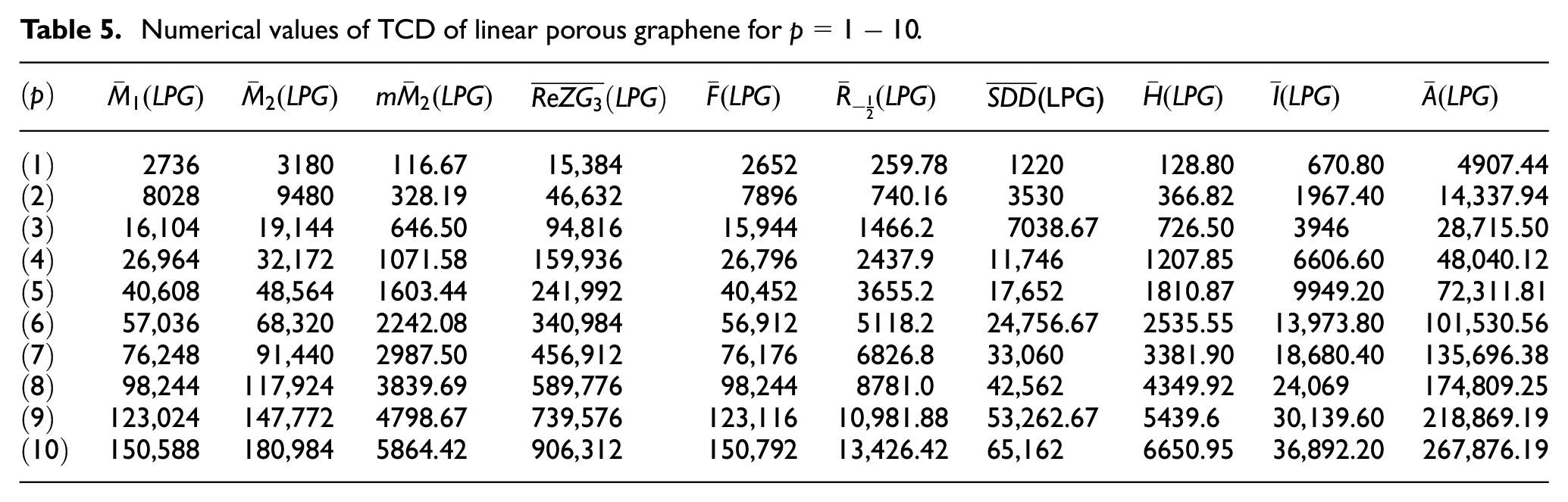
With the help of Table 1 we get the following and Table 5,
Numerical values of TCD of linear porous graphene for
CoM polynomial of triangular porous graphene (TPG)



Molecular graphs G
such tha
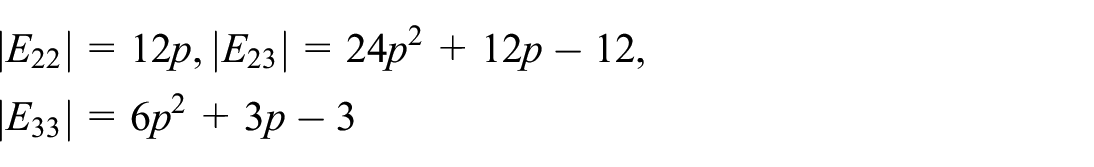
and, TPG’s vertex set classified into two classes as,

Using (1), we have


Similarly,
By definition of CoM Polynomial


1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.

then
Based on Table 1, we can find (Table 6)
Numerical values of TCD of triangular porous graphene for

Discussion
The calculation methods used in this study, namely the CoM Polynomial computations and curvilinear regression analysis, should be compared with other methods in the literature to benchmark their performance and highlight their advantages and limitations.
One method for comparison is density functional theory (DFT) calculations, which have been widely used to study lithium clusters. While DFT calculations provide accurate results, they are computationally expensive and limited to small cluster sizes. In contrast, the CoM Polynomial computations offer a more efficient and scalable approach for larger clusters.
Another method for comparison is machine learning-based approaches, such as neural networks and Gaussian process regression. These methods have been applied to predict cluster energies and properties, but require large amounts of training data and can be prone to overfitting. The curvilinear regression analysis used in this study offers a more interpretable and robust approach, especially when combined with the CoM Polynomial computations.
Additionally, other computational methods such as Monte Carlo simulations and molecular dynamics simulations can also be used to study lithium clusters. These methods can provide insights into the dynamic behavior and thermodynamic properties of the clusters, but are often limited by their computational cost and scalability.
A detailed comparison of these methods would require a thorough analysis of their accuracy, computational cost, and applicability to different cluster sizes and environments. This would provide a comprehensive understanding of the strengths and limitations of each method and highlight the advantages of the CoM Polynomial computations and curvilinear regression analysis used in this study.
By comparing the calculation methods used in this study with other methods in the literature, we can gain a deeper understanding of the complex behavior of lithium clusters and develop more accurate and efficient computational tools for predicting their properties and behavior.
Conclusions
Designing materials whose desired properties lead to the discovery of new structures has become increasingly difficult without theoretical guidance. Identifying key descriptors describing the structure of a material with the target property is the first step in this process. Therefore in this article, by utilizing codescriptors derived from CoM Polynomial and employing curvilinear regression analysis, we have theoretically predicted cluster energy of Lithium Cluster and we can summarize the results obtained in this article as follows:
Among ten codescriptors, Symmetric Division
For the studied property of
We derived generalized analytical expressions for degree-based codescriptors using the CoM Polynomial method applied to Linear and Triangular Porous Graphene structures. Additionally, we present graphical comparisons of these codescriptors for each structure, allowing for insightful visualization and analysis (Figures 10 and 11). Future research directions are illustrated in Figure 12.

Graphical visualization of Table 5.

Graphical visualization of Table 6.

Future work.
Footnotes
Handling editor: Sharmili Pandian
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The author(s) gratefully acknowledge Qassim University, represented by the “Deanship of Scientific Research, on the financial support for this research under the number (ENUC-2022-1-2-J-29899) during the academic year 1444 AH/2022 AD.”
Data availability statement
In this paper, the authors provided all raw data to support their conclusions.