
Abstract
Dyslipidemia is defined as elevated fasting blood levels of total cholesterol (TC), and its primary lipoprotein carrier—low-density lipoprotein (LDL), triglycerides (TG), or reduced high-density lipoprotein (HDL), alone, or in combination (mixed dyslipidemia). Dyslipidemia is well known to be associated with cardiovascular disease (CVD) risk. All patients with dyslipidemia should initiate therapeutic lifestyle changes to target lifestyle-related factors such as physical inactivity, dietary habits, and obesity. The combination of a proper dietary plan and regular aerobic exercise has been reported to lower TC, LDL-C, and TG by 7% to 18%, while increasing HDL-C by 2% to 18%. Numerous pharmacological therapies are available and aggressive therapy using a HMG-CoA reductase (3-hydroxy-3-methyl-glutaryl coenzyme A reductase) inhibitor (statins) should be initiated if lifestyle therapy is not enough to achieve optimal lipid levels with a primary target of lowering LDL-C levels. Aggressive treatment of dyslipidemia with maximal dosage of statin drugs have been reported to reduce LDL-C by 30% to 60%. If mixed dyslipidemia is present, a combination therapy with statin, niacin, cholestyramine, or fibrates should be initiated to reduce the risk of CVD events. These strategies have been shown to reduce CVD risk and optimize LDL-C levels in primary and secondary prevention of CVD.
‘The evidence supporting reduction of CVD [cardiovascular disease] events by lowering LDL-C and TC is strong and therefore should be considered primary therapeutic targets.’
Background
A high prevalence of dyslipidemia, defined as elevated fasting blood levels of total cholesterol (TC), low-density lipoprotein, (LDL), triglycerides (TG), and/or a reduced level of high-density lipoprotein (HDL), alone, or in combination, exists in the United States.1,2 In particular, 98.9 million adults or 43.4% of the US population have a TC >200 mg/dL, 31.1% have an elevated LDL-C level, and 21.8% have a reduced HDL-C level.1,2 Although lipids contribute to many vital functions in the body, dyslipidemia (especially elevated LDL-C levels) contribute significantly to the pathophysiology of atherosclerosis and the development and progression of coronary heart disease (CHD) beginning during the first decades of life. Dyslipidemias are heavily influenced by lifestyle choices and environmental factors, primarily via an inheritance mode that is polygenic. Familial hypercholesterolemia (FH), which is the result of a number of inherited autosomal dominant monogenic alterations to genes coding the LDL receptor and apoplipoprotein B (ApoB),3,4 is less common and will not be discussed in detail. FH is defined by the National Lipid Association “as a group of genetic defects that is characterized by a severe elevation of serum cholesterol levels.” 5 It should be noted that there are other causes of inherited elevated cholesterol levels, and thus FH is not limited to autosomal dominant FH.5,6 Treatment for FH includes aggressive lipid-lowering pharmacotherapy and lifestyle modification. Statin drugs and cholesterol absorption inhibitors (eg, ezetimibe) are the primary pharmacological agents for FH, with some patients requiring more intense therapies such as LDL-apheresis. 6 The reader is directed to the National Lipid Association Guidelines for FH for more information. 5
Primary and secondary prevention of CHD and other cardiovascular diseases (CVDs) include management of blood lipid levels via lifestyle and pharmacological therapies. The reader should note that, at the time this article is written, the National Cholesterol Education Program Adult Treatment Panel IV (ATP) is in advisory council for review and has not yet been published. Thus, recommendations in this article are based on the 2004 published ATP III guidelines and are subject to change on publication of ATP IV. Table 1 show the ATP III blood lipid and lipoprotein classifications.7,8 The purpose of this article is to provide an update on recent guidelines and trials on the treatment of dyslipidemia with a specific focus on lifestyle medicine.
Adult Treatment Panel III Blood Lipid and Lipoprotein Classification. a

Abbreviations: LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol.
Lipid classification based on values after a 12-hour or longer fast and the values shown are in mg/dL. Values for non-HDL-C are 30 mg/dL higher than the value for LDL-C. To convert mg/dL to international unit mmol/L divide cholesterol values by 38.7 and triglycerides by 88.54.
Adapted from Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. 7
Elevated Total and LDL Cholesterol
Over the past 50 years, numerous large-scale longitudinal, observational studies,9-11 have reported a strong positive association between TC, and LDL-C levels and CHD risk (ie, following adjustment for other major CHD risk factors there is a 2% increase in CHD risk for every 1% increase in TC >180 mg/dL). 11 Apo B100 concentration appears to be an even stronger predictor of CHD than LDL-C.12,13 There also have been a number of studies that have shown an association of lipoprotein-α with increased risk of CHD. 12 These observational studies have been supported by numerous randomized, controlled clinical trials successfully reducing CHD morbidity and mortality using cholesterol-lowering drugs such as 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase inhibitors (statins), cholestyramine, niacin, and fibrates. 14 The evidence supporting reduction of CVD events by lowering LDL-C and TC is strong and therefore should be considered primary therapeutic targets.
Statin drugs are the preferred initial pharmacological therapy for the treatment of dyslipidemia and aggressive lowering of LDL-C and TC. Statin drugs have as a class been shown in several recent meta-analyses to significantly reduce CHD and all-cause mortality in primary prevention populations.15-18 This apparent success of statin drugs in primary prevention of CHD related mortalities15-18 has led to the speculation that initiation of statin drugs earlier in the lifespan may prevent or forestall the onset of atherosclerotic CHD, albeit this remains a speculation as no randomized controlled trial has investigated this to date.19,20 Despite the clinically relevant, pleiotrophic effects of statin drugs on cardiovascular risk factors, it is apparent that reaching optimal LDL-C levels as recommended by ATP III is challenging. Several epidemiological studies21 -23 evaluating lipid-modifying agents, particularly statin drugs, have reported a wide variation (18-77%) in reaching the current ATP III target treatment goal of an LDL-C level of ≤100mg/dL in high-risk patients (ie, those with a history of CHD or CHD risk equivalent).24-26 Achieving an ATP III optional target treatment goal of an LDL-C level of ≤70 mg/dL in high-risk patients was reported to be even lower at 15% to 30%.24,26 The ATP III currently recommends initiating statin therapy if lifestyle therapy has failed to achieve optimal LDL-C levels, with an initial goal of reducing LDL-C levels ≥30% to 40%. More aggressive treatment with higher dosages or with the use of combination treatment using other lipid modifying pharmaceuticals may be needed to achieve optimal LDL-C levels. Table 2 lists the recommendations for initiation of statin drug treatment and LDL-C reduction goal.
Adult Treatment Panel III Recommendations for Statin Drug Initiation and LDL-C Reductions. a
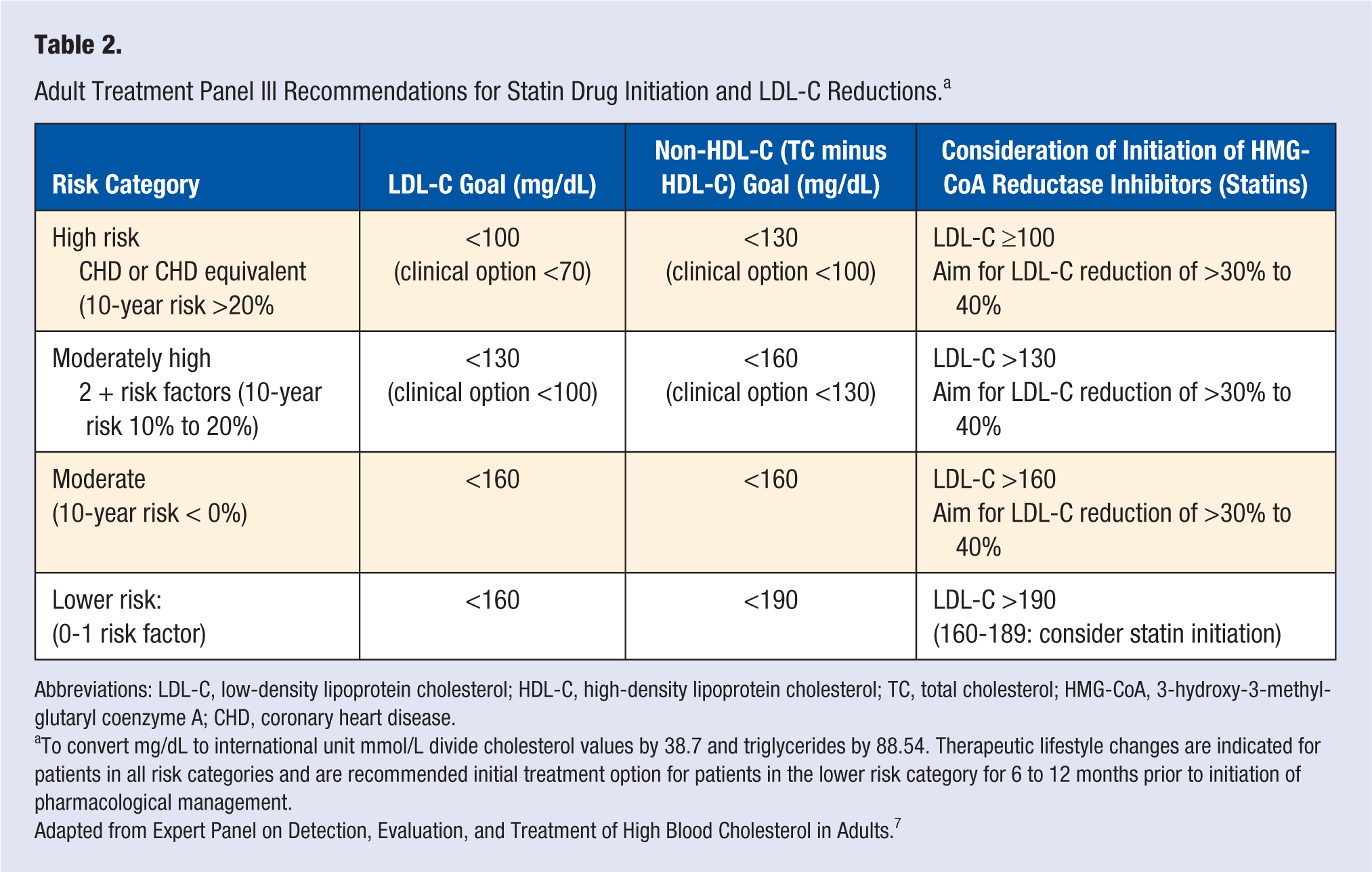
Abbreviations: LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol; TC, total cholesterol; HMG-CoA, 3-hydroxy-3-methyl-glutaryl coenzyme A; CHD, coronary heart disease.
To convert mg/dL to international unit mmol/L divide cholesterol values by 38.7 and triglycerides by 88.54. Therapeutic lifestyle changes are indicated for patients in all risk categories and are recommended initial treatment option for patients in the lower risk category for 6 to 12 months prior to initiation of pharmacological management.
Adapted from Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. 7
Potential Adverse Effects of Statin Drugs
In 2012, a black box warning was placed on statin drugs, primarily because of the reported effects on memory loss and confusion. 27 The Food and Drug Administration (FDA) also cautions that statin drugs may result in an elevation of blood glucose levels and increased risk of type 2 diabetes mellitus. 27 Additionally, medications to treat HIV, and bacterial and fungal infections may interact with certain statin drugs, increasing the risk of myopathies or rhabdomyolysis. 28 Other meta-analyses29,30 have reported a slight increase in the risk for diabetes development, but no evidence of an increased risk of cancer.15,30 It should be noted that the increased risk of myopathies, rhabdomyolysis, or elevated blood glucose levels appear to be mainly in those who receive aggressive statin therapy, 29 and the benefits of statin drugs in reducing cardiovascular events currently outweighs the potential risks associated with statin drugs. 30
High-Density Lipoprotein
A reduced plasma level of HDL-C has been shown to be inversely associated with atherogenesis and CHD in a number of large prospective studies and is generally considered a secondary pharmacotheraputic target. 31 A high HDL-C level appears to have several cardioprotective effects against CHD as demonstrated by numerous randomized, controlled trials through different pathways including: cholesterol transport from foam cells to the liver, improved endothelial function (likely due to an antioxidant and anti-inflammatory effect of HDL-C) with an apparent reduction in endothelial-derived adhesive proteins, and possible antithrombotic and profibrinolytic effects. Some studies have even reported a small regression of atherosclerotic plaque in response to aggressive HDL treatment.32-35 The general consensus is that the main antiatherosclerotic function of HDL lies within its ability to remove cholesterol from macrophages within the atheroma and transport it to the liver for disposal in the bile as cholesterol or as bile acids. 36 Over the past decade, much research has focused on therapies to increase both total and functional HDL-C levels. Degoma and Rader 37 have categorized these HDL-directed pharmacotherapeutic strategies into 4 divisions: indirect augmenters of apo A-I (the primary apolipoprotein of HDL-C) and HDL-C (cholesteryl ester transfer protein [CETP]) inhibitors, direct augmenters of apo A-I (intravenous infusion of recombinant or purified native apo A-I), apo A-I mimetic peptides, and enhancers of reverse cholesterol transport (lecithin:cholesterol acyltransferase [LCAT] activators, and liver X and farnesyl receptor agonists).
Indirect methods of enhancing apo A-I and HDL-C, appear to be the current foci of HDL therapies. CETP is the primary enzyme target of such pharmacological interventions. CETP is a plasma glycoprotein that is bound to HDL particles and is essential in the transfer of cholesterol ester from HDL to apo-B lipoproteins such as LDL, intermediate-density lipoprotein (IDL), and very low density lipoprotein (VLDL) in exchange for triacylglycerol. 38 Specifically, CETP forms a ternary complex with HDL and apo-B lipoproteins and forms a passageway through which cholesterol ester and TG are transferred. 39 If CETP is inhibited via pharmacological measures or inherited, there is a resultant increase in HDL-C levels postulated to result in a reduction in atherogenesis. Such pharmacological agents have shown promising effects on elevation of HDL-C, although significant heterogeneity exists with respect to size, and lipid and protein content. Moreover, this enzyme may have both pro- and antiatherogenic effects. Proatherogenic effects reflect the ability of CETP to increase cholesterol ester transfer to apo-B containing lipoproteins, which culminates in atherogenic uptake of cholesterol ester by macrophages present in the vascular wall. 39 Antiatherosclerotic effects of CETP are attributed to its effects on elevating circulating levels of HDL, possibly as a result of enlargement of existing HDL particles due to inhibition of core lipid exchange with VLDL and LDL particles. 39 In addition, there appears to be an increase in apo-A-I containing lipoproteins. 40 Inhibition of CETP by pharmacological agents such as Torcetrapib, Dorcetrapib, Anacetrapib, and Evacetrapib have been investigated as therapeutic agents for elevation of HDL.40-51
The first phase III trials investigating the inhibition of CETP by the drug Torcetrapib (ILLUSTRATE, RADIANCE 1 and 2, and ILLUMINATE trials) did indeed result in increased serum HDL levels, 51% to 72%41-44; however, the elevation in HDL levels did not reduce progression of atherosclerosis as measured by carotid intima media thickness. 45 Furthermore, Torcetrapib was associated with a significant pressor effect46,47 and increased risk of cardiovascular events 41 and trials were discontinued. The failure of Torcetrapib to reduce cardiovascular endpoints, despite its HDL elevating effects, suggest that the functionality of HDL (indicated by the presence of apolipoproteins apo-A1 and apo-E, and enzymes paraoxonase-1 and acetylhydrolase), is an essential component for cardioprotective effects of HDL, rather than serum levels alone, as discussed by Zheng and Aikawa. 39 Recent CETP phase IIb trials (dal-VESSEL and dal-PLAQUE) for Dalcetrapib were shown to lack the pressor effects of Torcetrapib,48,49 but ultimately proved inadequate in promoting a strong lipid altering effect. Dalcetrapib appears to improve in HDL by 31% and TG (2.6% to 7.0%), with minimal effects on LDL-C.48,49 Dalcetrapib did not improve endothelial function, 49 reduce vessel wall inflammation, 48 or slow progression of atherosclerosis. 48 The phase III trial (dal-OUTCOMES) for Dalcetrapib was discontinued because of a lack of meaningful clinical effects. 51 The most current CETP inhibitor Anacetrapib has passed phase II trials, and has shown greater HDL altering affects than Torcetrapib, with the apparent lack of side effects. Although no hard clinical endpoints were evaluated, the DEFINE trial reported improvements of HDL of 138%, as well as a lowering of LDL and TG by 40% and 5% respectively. 50 Additionally, it appears that Anacetrapib does not impair the ability of HDL to suppress inflammatory cytokines and adhesion molecules, 52 which suggests that the altered HDL may be of a functional phenotype. Results of the phase III clinical trial REVEAL are scheduled to be published in 2017, which will help establish the potential clinical benefit of Anacetrapib. 53
Although a large amount of HDL related therapies have focused on CETP inhibition, agents without the concerned toxicities are currently being developed. Several of the innovative HDL-promoting therapies currently in various stages of development include apo A-1–related peptides, endothelial lipase inhibitors, 54 nuclear hormone receptor agonists that target the dual peroxisome proliferator–activated receptor (particularly for type 2 diabetes),55,56 liver X receptor,57,58 and farnesoid X receptors.57,58 One promising therapy, Aleglitazar, a dual peroxisome proliferator–activated receptor agonist, produced HDL increases upward of 20% while simultaneously significantly reducing LDL, TG, and apo B levels in the phase II trial SYCHRONY. 56 However, no preliminary data have, to our knowledge, been published from phase III trials in regard to Aleglitzar, and the other HDL-altering therapies discussed above.
Niacin is one of the few HDL cholesterol raising therapeutic agents currently available and is regularly prescribed for its HDL-altering effects.59-61 Niacin’s primary mechanisms of actions include increases in HDL-apo-A1 particles and HDL biogenesis by inhibiting hepatocyte surface expression of β-chain adenosine triphosphate synthase and increased expression of hepatic expression of adenosine triphosphate–binding cassette transporter-1 (ABCA1), respectively. 62 As with statins, niacin exhibits multiple pleiotropic, lipid independent effects, including the inhibition of vascular endothelial oxidative and inflammatory events, inhibition of neutrophil myeloperoxidase, and formation of dysfunctional HDL, all postulated factors in the progression of atherosclerosis. 62 Recent research had focused on the use of the combination of niacin and other cholesterol lowering drugs (eg, statin drugs). For example, the “Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides: Impact on Global Health Outcomes” study (AIM HIGH) 63 investigated the role of dual niacin/simvastatin therapy over simavastatin therapy alone, in patients with CVD. Results of the trial appear to be disappointing, as patients receiving dual niacin/statin treatment saw no reduction in cardiovascular events compared with those receiving statins as a monotherapy. 63 However, the authors note that caution should be used when interpreting these findings, due to the strong effectiveness of statin therapy in patients prior to trial enrollment. 64 Because of the success of niacin as a monotherapy in previous clinical trials, niacin continous to be a widely used and successful treatment for the prevention of cardiovascular events. 65
Triglycerides
Epidemiological studies have reported an association between elevated TG and CHD (eg, following adjustment for CHD risk factors; at TG level >88.5 mg/dL, there is an associated increase in CHD risk by 37% and 14% for women and men, respectively, for every 1 mmol/L increase in TG level).66,67 However, hypertriglyceridemia does not appear to be as strong a CHD risk factor as LDL-C. This is in part because of the constellation of CHD risk factors commonly associated with elevated TG levels as evidenced by an attenuation or even loss of association between TG levels and CHD following adjustment for multiple CHD risk factors. Clinicians should note that there is a greater biological, and laboratory measurement variability of TG levels compared with the other commonly measured blood lipids. 68 It is currently conjectured that nonfasting TG levels may provide a more predictive assessment of CHD risk, since postprandial TG remnant lipoproteins can cross the vascular endothelial cell barrier and thereby deliver TG substrate for foam cell development.69-71 It should be noted that there have not been any clinical intervention trials that have specifically targeted TG levels alone. However, subgroup analyses of trials involving fibrate or statin drugs have indicated a reduction in CHD incidence of ~1% for each 1% reduction in TG level.70,71
Non-HDL Cholesterol and Apo B
Non-HDL-C (calculated by deducting HDL-C from TC levels) has been reported to be a better predictor of CHD than LDL-C or HDL-C alone, especially in patients with the metabolic syndrome, overt diabetes, or kidney disease. 72 This is conjectured to be related to plasma apo B level, which represents the total atherogenic particle number in plasma (ie, VLDL + IDL + LDL), since LDL, IDL, VLDL and their remnants each contain 1 molecule of apo B following TG lipolysis.72-74 Therefore, the ATP III currently suggests that the concentration of cholesterol that is contained within all apo B-containing lipoprotein particles (ie, non-HDL-C) should be considered a secondary pharmacotheraputic goal in the presence of an elevated TG level with a primary goal of reaching the target treatment goal of LDL-C (Table 2).7,8
Lipid Classification and Treatment Targets
The clinical fasting blood lipid profile consists of TC, HDL-C, and TG for assessment of CVD risk. LDL-C is most commonly estimated by the Friedewald formula of TC − TG/5 = LDL (mg/dL). 75 There is an assumption of a constant cholesterol/TG ratio of VLDL; however, if the TG values are >400 mg/dL, this formula cannot be used. In this case, isolation of LDL fraction and direct quantification of LDL-C content is required. Patients with dyslipidemia are stratified and treatment target goals are based on their relative risk of experiencing a major CVD event. Clinicians should be aware of the possibility of misleading TC values when assessing CVD risk, especially in women with high HDL-C levels. It is recommended to use both LDL-C and HDL-C in CVD risk stratification (this does not include patients with FH or hypertriglyceridemia, which warrants special attention as a high CVD risk population). Clinicians also should note that patients that present with a condition that is known as a CHD equivalent or other CVD diagnosis are stratified into the high-risk group.
Patients are often classified based on their 10-year risk as derived from the Framingham Risk Score as indicated by ATP III 7 and American Heart Association (AHA) guidelines. 76 This classification allows for stratification and level of intensity of treatment. Patients should be classified into high risk (10-year CVD risk >20%), moderately high risk (10-year CVD risk 10% to 20%), moderate risk (10-year CVD risk <10%), or low risk (0-1 CVD risk factors).60,61 Patients presenting with an elevated LDL-C level should be screened and treated for any additional major CVD risk factors (eg, cigarette smoking and hypertension).77,78 Clinical judgment should used to upgrade a patient to a higher risk category and aggressiveness of treatment should be based on availability on clinical findings and medical history. It should be noted that other validated risk evaluations such as the Systematic Coronary Risk Evaluation (SCORE) 79 are becoming increasingly common, particularly in Europe, and clinicians should use clinical judgment when using different risk evaluation scores available. 80 Table 2 lists by risk category status, LDL levels recommended for initiating “therapeutic lifestyle changes,” and for the “consideration of drug therapy,” and the associated LDL-lowering goals.
All patients should initiate therapeutic lifestyle changes to target lifestyle-related risk factors (physical inactivity, dietary habits, and obesity). If the target treatment level is not achieved within 6 to 12 months, pharmacological intervention is warranted, preferably via initiation of statin drug treatment with the goal of achieving 30% to 40% reduction in LDL-C levels to achieve the greatest clinical benefit. Clinicians should be aware of the different available statin drugs and statin based therapies that currently are available for use to achieve on-treatment target LDL-C level. If the initial statin drug treatment fails to achieve a target LDL-C level, intensification of treatment by increasing dosage or consider using a combination therapy. 81 Patients presenting with elevated TG or low HDL-C levels many benefit from a combination of statin therapy, and a fibrate drug or nicotinic acid, or fish oil supplement (1-4 g/d) as recommended by ATP III guidelines. Table 3 summarizes the usual percentage changes in blood lipid/lipoprotein levels with each specific class of drugs. For more details and precautions in use of these lipid management pharmaceuticals the reader is referred to Bettridge and Morell. 82 Readers should be advised that increasing statin dosage or combining a statin with other therapies may also raise the risk of myopathies such as myalgia or myositis.28,83
Percentage Change in Blood Lipid/Lipoprotein Levels Corresponding to Specific Class of Drugs. a
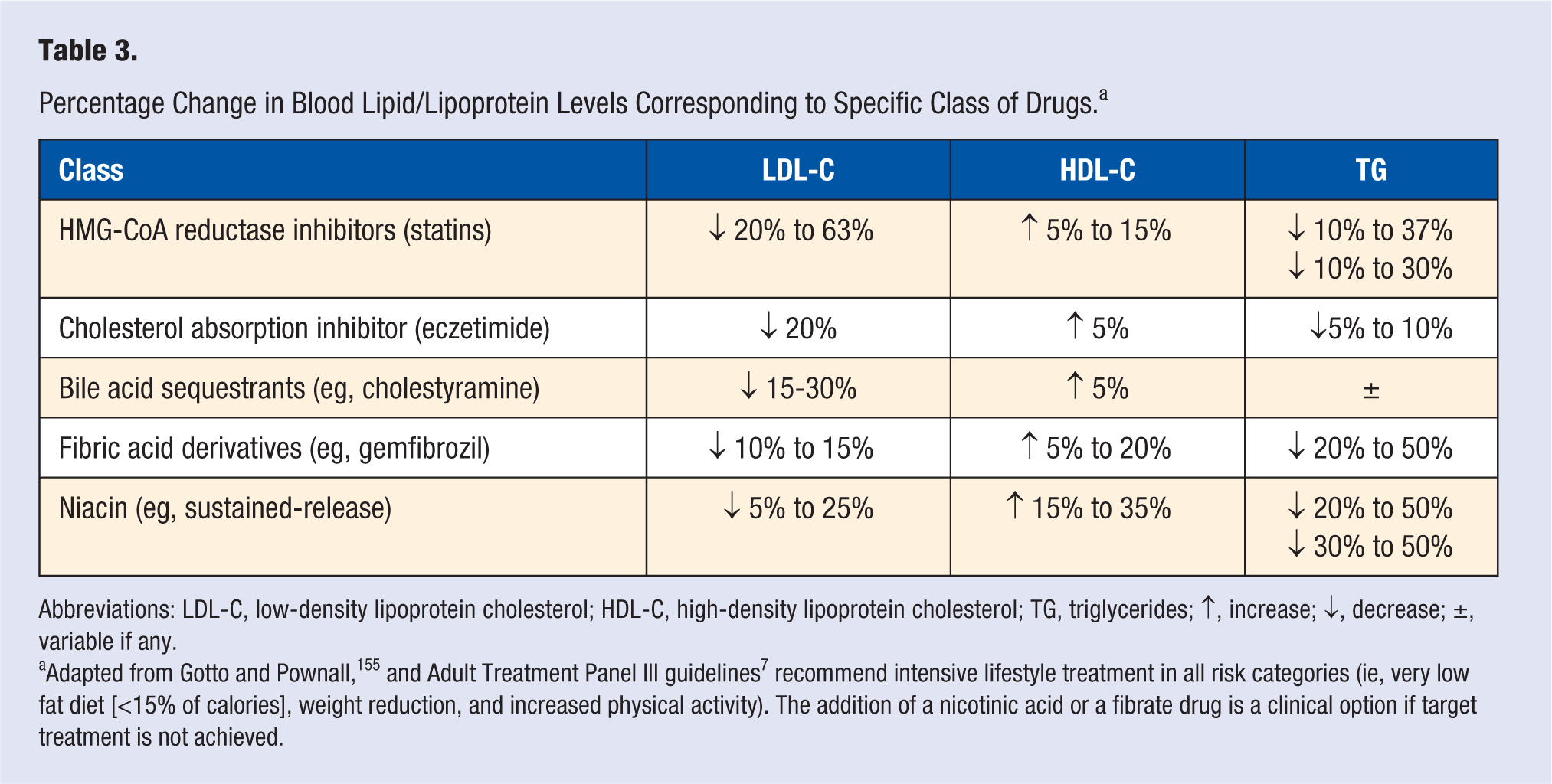
Abbreviations: LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol; TG, triglycerides; ↑, increase; ↓, decrease; ±, variable if any.
Adapted from Gotto and Pownall, 155 and Adult Treatment Panel III guidelines 7 recommend intensive lifestyle treatment in all risk categories (ie, very low fat diet [<15% of calories], weight reduction, and increased physical activity). The addition of a nicotinic acid or a fibrate drug is a clinical option if target treatment is not achieved.
Role of Lifestyle in the Management of Dyslipidemia
Therapeutic lifestyle change (dietary habits, body weight reduction, physical activity, moderation of alcohol consumption, and smoking cessation) is considered of primary importance and lifestyle habits are primary targets for the management of dyslipidemia.
Impact of Diet
Randomized, controlled, clinical trials of cardiometabolic risk factors (including dyslipidemia) and prospective cohort studies provide overwhelming evidence of cardiovascular benefits of several specific foods. 84 Although individual nutrients in isolation may alter cardiometabolic risk factors, the health effects of nutrients likely act synergistically, and may be especially potent when consumed as part of a prudent dietary pattern, such as the Mediterranean, DASH (Dietary Approaches to Stop Hypertension), or vegetarian style diets. 85 Such longevity promoting dietary patterns, particularly the Mediterranean diet, have well-established cardioprotective benefits, partially mediated through improvements in dyslipidemia.86-89 Specifically, a meta-analysis of 50 studies (15 epidemiological and 35 randomized control trials) provides substantial evidence that adherence to the Mediterranean diet significantly improved all aspects of the metabolic syndrome such as TG and HDL-C. 90 Additionally, research by Sola et al 91 shows that individuals classified as high cardiovascular risk who followed a Mediterranean dietary pattern rich in virgin olive oil, reduced their apo B and apo B/apo A-I ratio and improved apo A-I concentrations. Table 4 summarizes the effects of specific dietary components and other therapeutic lifestyle changes reviewed in this article on dyslipidemia.
Effect of Dietary Components and Other Therapeutic Lifestyle Change on Dyslipidemia.
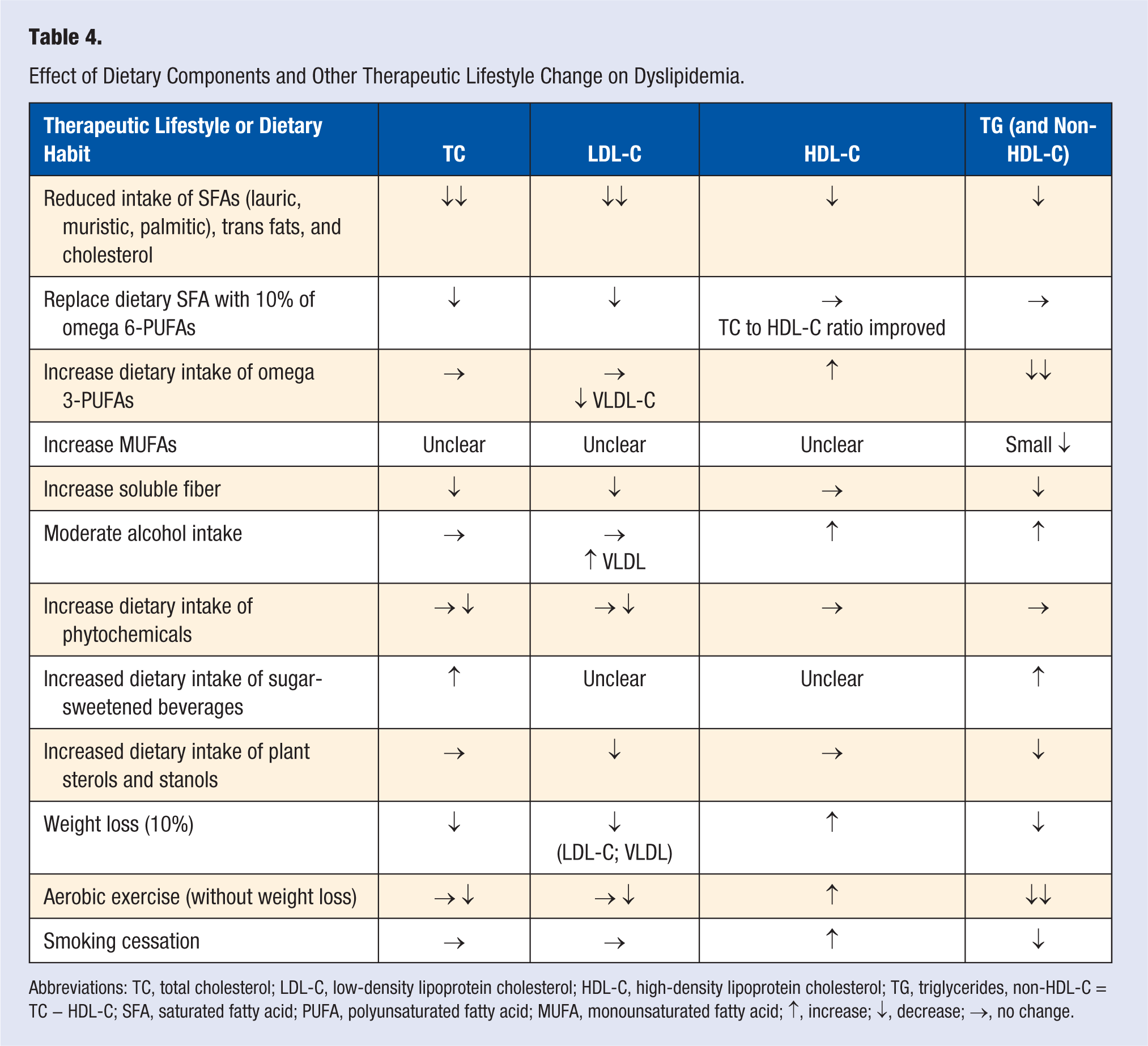
Abbreviations: TC, total cholesterol; LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol; TG, triglycerides, non-HDL-C = TC − HDL-C; SFA, saturated fatty acid; PUFA, polyunsaturated fatty acid; MUFA, monounsaturated fatty acid; ↑, increase; ↓, decrease; →, no change.
The primary components included in a prudent dietary pattern, intended to reduce dyslipidemia, focus on fat quality with reduced intake of animal food sources, specifically red meat, dairy products, butter, hard margarines, and commercial baked goods. 92 These foods contain saturated fatty acids (SFAs) that are known to contribute significantly to elevated blood lipid/lipoprotein levels (ie, the 3 long-carbon chained SFAs responsible for increasing TC; lauric [12:0], muristic [14:0], and palmitic [16:0] acids) by downregulating hepatic synthesis of LDL apo B/E cell receptors, which increases levels of LDL, VLDL, IDL, and apo B-100 remnants.93,94 The observation of the association between increased consumption of SFA, TC, and CHD has been confirmed over the past decades in multiple observational studies and controlled intervention trials.88,92,95 The importance of reducing dietary SFA intake is reflected by Keys’s equation indicating that for every 1% reduction in SFAs, plasma TC is reduced 2%. 96 Clinicians should note that stearic acid (18:0) and SFAs with 10 or less carbons appears to have no effect on TC, and dietary cholesterol intake exerts a relatively small impact on TC plasma levels since two thirds of circulating cholesterol is synthesized de novo by the liver from acetyl coenzyme A products of metabolism thereby limiting its absorption. 93
Trans Fatty Acids
Essentially all of the 2.6% of daily energy intake of trans fatty acids (TFAs) results from intake of margarines and shortenings used in food preparation and only a very limited quantity of TFAs occurs naturally in meat and dairy products. TFAs promotes an increase in cholesterol synthesis and a concomitant increase in LDL-C levels, while in contrast to SFAs, they also increase lipoprotein(a) and lower HDL-C levels. 97 Therefore, restriction of dietary trans fatty acids intake should be obligatory, or preferably eliminated. Currently, it is recommended that dietary intake is limited to <1% of total energy intake. 98
Omega-6 Polyunsaturated Fatty Acids
Epidemiologic evidence suggests that the replacement of 10% of calories from SFA with linoleic acid (omega-6 polyunsaturated fatty acids [PUFAs]), is associated with an 18 mg/dL reduction in LDL-C. 99 However, higher plasma PUFA levels are often associated with reduced TC to HDL-C ratio. These findings are reflected in the current ATP III guidelines to keep intake of omega-6 PUFAs to 10% or less of daily energy intake. 7 Omega-6 linoleic acid (18:2, n-6) is found in sunflower, corn, and soybean oils, seeds, and nuts, and appears to increase removal of lipoproteins and reduce conversion rate of VLDL to LDL by upregulating hepatic LDL receptors. Linoleic acid also is important in the formation of arachadonic acid (20:4, n-6) and production of prostaglandins and eicosanoids. 36
Omega-3 Polyunsaturated Fatty Acids
Omega-3 PUFAs (long-chained, essential fatty acids) are required in the diet and are postulated to contribute to primary and secondary prevention of CHD.94,100 The omega-3 PUFAs, eicosapentaenoic acid (EPA; 20:5, n-3) and docosahexaenoic acid (DHA; 22:5, n-3), are derived from the parent compound omega-3 PUFA α-linolenic acid (ALA; 18:3, n-3), and are believed to be cardioprotective.100-104 ALA is present in vegetable oils, (eg, canola oil), whereas fatty fish such as salmon, mackerel, herring, and sardines are rich sources of EPA and DHA.
Epidemiological observational studies have consistently reported an inverse association between consumption of fish or fish oil supplements with CVD morbidity and mortality. 105 Although not demonstrated by all primary and secondary prevention trials,106-108 the consensus gathered from the majority of randomized controlled trials (and meta analyses of randomized controlled trials) support the notion of reduced cardiovascular related mortality with adequate daily consumption these fatty acids.102,109-112 Additionally, the latest estimate of CHD-related mortality reduction for those who consume 250 mg/d of EPA/DHA, the current amount recommended in the latest 2010 Dietary Guidelines of America, 98 is 36%. 113 It appears that a modest intake of fatty fish, that is, 2 to 3 portions per week or a daily fish oil capsule can reduce risk of CHD, whereas consumption of ALA from vegetable oils does not appear to be cardioprotective. A review and meta-analysis on the topic by Pan et al 104 suggests that for each 1 g/d incremental intake of ALA there is an associated 10% reduction in CHD-related death.
Docosahexaenoic acid and EPA fish oil–based supplements appear to result in a reduction in TG (5% to 10% per gram of EPA and DHA), VLDL-C, VLDL-TG, and non-HDL-C, and an increase in HDL-C. 114 The combination of a statin drug and large doses of EPA-DHA based fish oil supplements appears to be an effective therapeutic approach for treatment of mixed dyslipidemia. 115 Specifically, the AHA recommends 2 to 4 g of EPA/DHA per day for patients who need to lower TG. 116 Clinicians should be aware that a dose–response relationship has been demonstrated for TG reduction, up to 7 g/d of EPA/DHA, and the response appears to be more pronounced in those with higher baseline levels and clinical judgment should be used to determine optimal treatment levels. 103
Monounsaturated Fatty Acids
Monounsaturated fatty acids (MUFAs) found in olive oil, canola and peanut oil, avocados, nuts, and fish appear to make LDL less susceptible to oxidation. It is currently unclear if MUFAs (eg, oleic acid found in olive oil) has an effect on blood lipid levels.117,118
Carbohydrate and Fiber Content
In addition to the focus on fat quality, carbohydrate quality is a staple of prudent dietary patterns and also has favorable effects on dyslipidemia. 84 Prudent dietary patterns are often considered rich sources of complex, fibrous carbohydrates, low in simple sugars (such as sugar-sweetened beverages), and has a low glycemic index, all of which have favorable factors on TG, TC, and LDL-C levels.119-121 Soluble fiber components (found in whole grains, fruits and vegetables, and beans and legumes) and in particular β-glycans and pectin, contribute to blood cholesterol reduction by binding to bile salts in the small intestine and thereby attenuating reabsorption. The attenuated reabsorption of bile salts leads to an upregulation of hepatic LDL receptors, which enhances LDL removal and subsequent reduction of plasma LDL-C levels. LDL-C levels (including the atherogenic small dense LDL particles) have been shown to be reduced by as much as 5 mg/dL following a regular dietary intake of 30 g/d of soluble fiber, potentially leading to a reduction in CHD events.122-126
Plant Sterols and Stanols
Plant steroids and stanols found in vegetable oils, seeds, and nuts 127 appear to reduce LDL-C and non HDL-C levels.127-129 This is postulated to be because of a competitive inhibition of biliary and dietary cholesterol, which leads to an up-regulation of hepatic LDL receptors. 92 Stanol-enriched margarines may be useful as an adjunct treatment to reduce plasma levels of LDL-C as endorsed by ATP III. Other plant-derived vitamins and antioxidants (eg, vitamins E and C and β-carotene) are postulated to improve endothelial function and prevent vascular oxidative stress and inflammation thereby reducing LDL oxidation and lessen CHD risk. 130 However, clinicians should be aware that multiple large-scale randomized controlled clinical trials have failed to demonstrate any CVD protective effect by supplementation of antioxidant vitamins (vitamin E, vitamin C, or β-carotene).130,131
Phytochemicals
Another type of plant chemicals (phytochemicals), found in fruits, vegetables, tea, red wine (and grape juice from which wine is derived), and cocoa bean, have shown the ability to improve several aspects of dyslipidemia.129,132-135 Phytochemicals such as soy isoflavones, catechins, resveratrol, monacolin, and lycopene have been demonstrated in both small scale clinical trials and in vitro studies to reduce cholesterol,133-135 and inhibit both LDL oxidation 136 and HMG-CoA reductase (although weakly).132,137,138 Because of the positive findings of several of the above mentioned trials, the role of micronutrients and phytochemicals in the treatment of dyslipidemia is an intense area of study by the neutraceutical industry. Although nutraceutical intervention (antioxidants and phytochemicals) may be an alternative for patients who are statin intolerant or refuse pharmacological intervention, the current evidence does not yet clearly support their independent use in a clinical setting.
Added Sugars (Sugar-Sweetened Beverages)
In the past 30 years, there has been a marked increase in the consumption of liquid calories, particularly in the form of sugar-sweetened beverages (SSBs), 139 that have been linked to increased cardiometabolic risk. 140 SSB promote weight gain and independently increase risk of cardiovascular diseases, type 2 diabetes mellitus, and metabolic syndrome due to the elevation of several risk factors, including atherogenic dyslipidemia. 140 SSBs contain varying concentrations of fructose and glucose in the form of high-fructose corn syrup, in which the former monosaccharide has been recently implicated as a key contributor to metabolic syndrome, obesity, and dyslipidemia.141,142 The increase in dyslipidemia is likely due to the promotion of de novo lipogenesis, apo B levels, and most notably fasting TG, which has been shown in several recent trials.143-147 However, the amount of fructose required to promote dyslipidemia is a source of debate. To address this question, Livesey and Taylor 148 performed a meta-analysis of 60 studies that evaluated the effects of fructose consumption on TG levels and observed that an intake of 100g/day of fructose resulted in a significant dose-related increase in plasma TG (an amount equivalent to 4 + 12 oz cans of cola). The findings of the meta-analysis agree with recent findings in a randomized controlled trial by Maersk et al 149 who reported that consumption of 1 L of cola per day for 6 months significantly increased TG (32%), TG (11.4%), and other components of the metabolic syndrome compared with consuming the same quantities of other liquids such as diet cola, milk, and water.
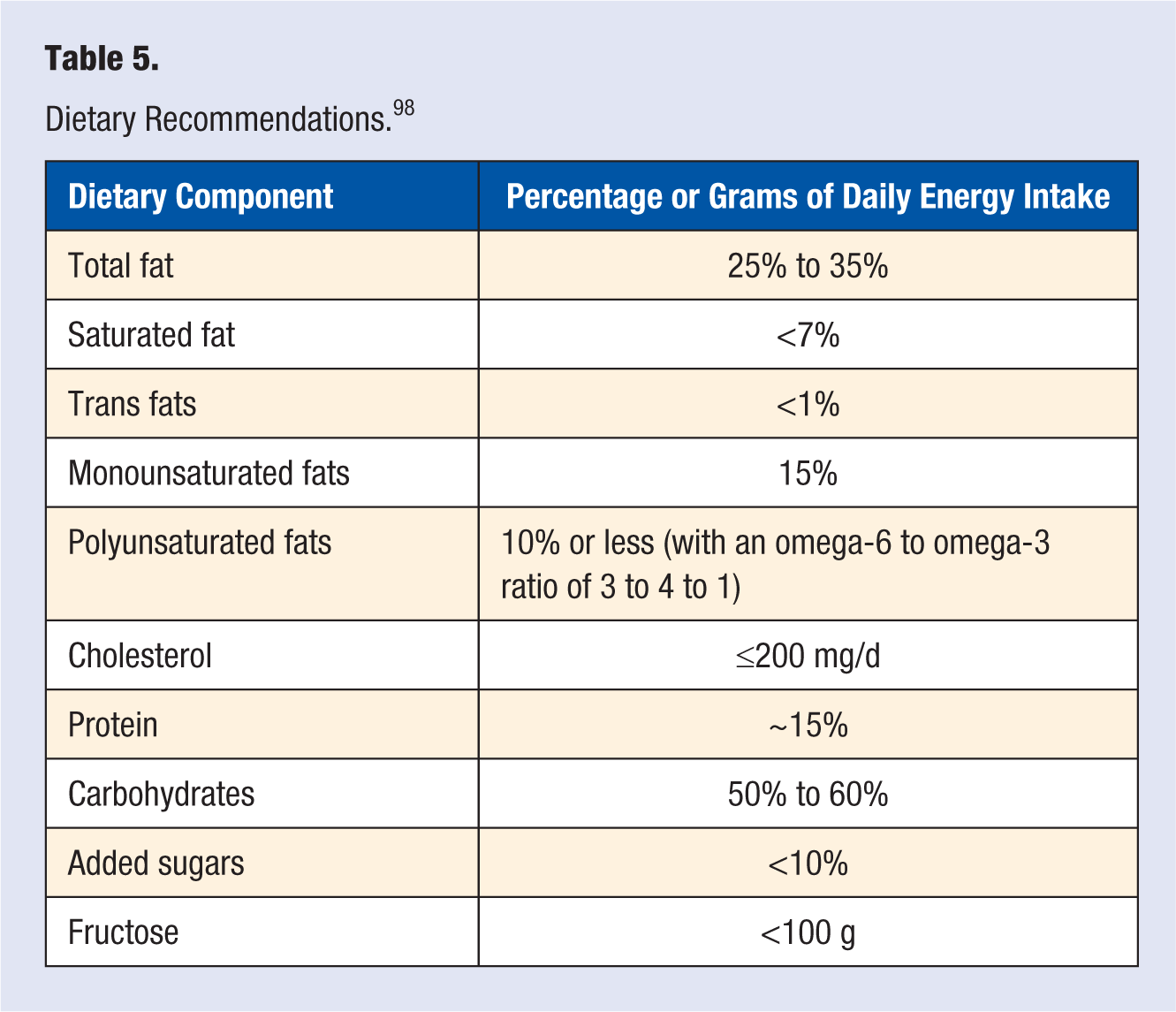
The implication of fructose, rather than glucose in dyslipidemia may stem from major differences in their metabolism. During a high energy state of the hepatocyte, there is a buildup of citrate and adenosine triphosphate, which inhibits glucose uptake and metabolism via negative feedback. This causes glucose to bypass the liver and enter systemic circulation for subsequent metabolism by other tissues. However, fructose metabolism is not inhibited, which allows fructose to undergo glycolysis and form metabolic intermediates that serve as a backbone for lipogenesis, particularly in times high cellular energy status. 150 Despite being a dense source of fructose, SSBs are also considered a high glycemic load, a high source of calories, and promote postprandial hyperinsulinemia. Hyperinsulinemia directly increases hepatic lipogenesis via an upregulation of sterol regulatory element-binding protein (SREBP-1c), a transcription factor that regulates fatty acid and TG synthesis. 151 Patients with dyslipidemia are therefore encouraged to refrain from consuming SSBs. Table 5 summarizes the current dietary recommendations by the Dietary Guidelines of America. 98
Dietary Recommendations. 98
Smoking and Alcohol
Cigarette smoking is known to be associated with an 11% to 14% reduction in apo A-I and HDL-C and a slightly higher LDL-C level, as compared with nonsmokers. This is postulated to be due to nicotine and carbon monoxide exposure.152-154 Successful cessation of smoking appears to increase HDL-C levels by 6 to 8 mg/dL and should be strongly encouraged in all patients. 155 Ethanol (moderate alcohol intake) appears to be associated with elevated levels of HDL-C by 12% 82 although clinicians should be aware that ethanol also increases TG and VLDL levels. ATP III does not currently recommend ethanol intake as a treatment for dyslipidemia and alcohol intake should be moderated, if alcohol is consumed. 7
Weight Management
Dyslipidemia is well known to be associated with obesity and weight loss is considered a cornerstone therapy in primary prevention strategies. The ATP III, 7 the AHA, 156 and the American College of Sports Medicine 157 recommend following a diet that results in a negative energy balance of 500 to 1000 kcal/d aiming for 1 to 2 lb (0.45-0.99 kg) weight loss per week while simultaneously ensuring that the daily energy intake is no lower than 1000 kcal/d, in order to provide all essential nutrients.156,157 Numerous dietary approaches have been reported for weight loss promotion and alteration of several cardiovascular risk factors. Such dietary interventions often focus on the alteration of macronutrient quantity and/or quality.158-162 Despite the success of many of these dietary approaches, it is apparent that the greatest impact on weight loss is the level of energy deficit promoted by the diet. It appears that a successful 10% weight loss may result in a significant decrease in TC of 30.5 mg/dL (−13.2%), LDL-C 15.1 mg/dL (−11.3%), VLDL-C 15.5 mg/dL (−36.5%), and TG 58.4 mg/dL (−32.2%); HDL-C initially declines during the active weight loss phase, and then increases 5.5 mg/dL (+12.6%) after weight stabilization. 146 This represents about a 0.35 mg/dL increase in HDL-C for every kg of weight lost. 163
Exercise
The effect of moderate- to high-intensity aerobic exercise for 30 to 60 minutes, 3 times per week with a weekly energy expenditure of 900 to 2000 kcal (or about 10 to 20 metabolic equivalent hours) for at least 12 weeks on dyslipidemia has been well documented.164-169 Aerobic exercise training results in a modest increase in HDL-C of 4% to 5% with an associated increase in apo A-I and LPL activity although genetic factors (and weight loss) contribute significantly to the exercise-induced changes in HDL-C.165,166,168,170-176 Few studies have been able to observe exercise-induced reductions in LDL-C and TC without sustained weight loss.165,166,168,170 Moderate-intensity exercise training appears to be effective in reducing TG levels (20% to 30% reduction) with an apparent dose–response relationship.168,177
The role of visceral fat (ie, a high accumulation of fat in the abdominal cavity) in dyslipidemia has become increasingly clearer during the past decade. 178 Studies that have used imaging techniques to assess the regional distribution of body fat have demonstrated that an excess of visceral adipose tissue is associated with a cluster of metabolic disturbances, including insulin resistance, hyperinsulinemia, glucose intolerance, hypertriglyceridemia, elevated apo B concentrations, small, dense LDL particles, as well as low HDL cholesterol levels. 178 Additionally, in patients with CHD, it has been shown that excess visceral adipose tissue is clearly the phenotype associated with the most severe atherogenic and diabetogenic abnormalities. 179 Anthropometric measurements such as waist circumference and waist to hip ratio are better indicators of mortality than body mass index alone in CHD patients, likely because of the associated cardiometabolic risk factors, including dyslipidemia.180,181 Therefore, it is essential to understand that abdominal adiposity exacerbates dyslipidemia and cardiometabolic risk 178 and visceral fat volume is likely to have a confounding effect on lipid changes with weight loss, diet, and exercise.
Conclusions and Recommendations
A large body of prospective epidemiological and randomized, controlled trials has provided robust evidence that dyslipidemia and successful management of dyslipidemia is crucial in the primary and secondary prevention of CHD. Therapeutic lifestyle change is an essential cornerstone treatment for all patients with dyslipidemia and should be initiated immediately on diagnosis of dyslipidemia. The combination of a proper dietary plan and regular aerobic exercise has been reported to lower TC, LDL-C, and TG by 7% to 18%, while increasing HDL-C by 2% to 18%. 182 Numerous pharmacological therapies are available and aggressive therapy using a statin drug should be initiated if lifestyle therapy is not enough to achieve optimal lipid levels with a primary target of lowering LDL-C levels. Aggressive treatment of dyslipidemia with maximal dosage of statin drugs have been reported to reduce LDL-C by 30% to 60%. If mixed dyslipidemia is present, a combination therapy should be initiated to reduce the risk of CVD events. These strategies have been shown to reduce CVD risk and optimize LDL-C levels in primary and secondary prevention of CHD and other CVDs.