
Abstract
Background
Extracellular matrix (ECM) deposition and excessive fibrosis are important factors in the deterioration of cardiac function after myocardial ischaemia‒reperfusion injury (I/RI). However, therapeutic strategies for inhibiting ECM deposition and excessive fibrosis have still not been elucidated.
Methods and results
Single-nucleus RNA sequencing (snRNA-seq) revealed that the overexpression of type VI collagen-α 3 (Col6a3) in fibroblasts in the myocardial infarction area strongly promotes the process of myocardial fibrosis. Consistent results were not observed in the infarcted myocardial tissues of mice treated with 4,8-dicarboxyl-8,9-iridoid-1-glycoside (BIG). Echocardiography confirmed that BIG alleviated cardiac dysfunction in mice after myocardial I/RI, TTC and Evans blue double staining revealed that BIG reduced the myocardial infarction size and area at risk. BIG inhibited inflammatory responses, apoptosis, and matrix metalloproteinase (MMP) secretion both in vivo and in vitro. Immunofluorescence staining revealed that BIG downregulated the expressions of TGF‒β and Col6a3 in cardiac fibroblasts but not in cardiomyocytes. The PI3K-specific inhibitor LY294002 and AKT inhibitor were utilitzed to confirm that BIG suppressed myocardial fibrosis and alleviated cardiac dysfunction by activating the PI3K/AKT pathway.
Conclusions
The results provide valuable information for the treatment of myocardial fibrosis induced by myocardial I/RI and highlight the therapeutic potential of BIG in reducing collagen deposition.

Keywords
Introduction
Myocardial ischaemia‒reperfusion injury (I/RI) represents a significant global health challenge, and existing therapeutic interventions are limited. 1 Cardiac fibroblasts are activated after myocardial I/RI, and activated fibroblasts secrete large amounts of fibrotic factors and extracellular matrix (ECM), which are deposited in the myocardial interstitium and perivasculature. 2 Virtually all instances of myocardial I/RI result in subsequent cardiac fibrosis, characterized primarily by fibronectin and collagen deposition in areas previously occupied by cardiomyocytes.3,4 Moderate fibrosis following myocardial I/RI plays a critical role in preserving the structural integrity of the heart. 5 However, excessive fibrosis results in increased ECM deposition and myocardial remodelling, ultimately leading to the progression of heart failure.6,7 Anticardiac fibrosis therapy is an effective intervention for reducing sudden death in patients subjected to myocardial I/RI. 8 Therefore, inhibiting cardiac fibrosis to prevent heart failure induced by myocardial I/RI is highly anticipated.
The activation of myocardial fibrosis after myocardial I/RI is mediated by various mechanisms, including the release of inflammatory cytokines, excessive replacement fibrosis, transforming growth factor‒β (TGF‒β) and increased ECM deposition. Nanobodies loaded with reactive oxygen species hydrogels targeted to the proinflammatory cytokine IL-1β ameliorate fibrosis in the myocardium subjected to I/RI and improve cardiac function. 9 Similarly, TGF-β1/SMAD3 plays a key role in mediating the process of cardiac fibrosis. 10 Type VI collagen (Col6) is deposited and accumulates in infarcted myocardial tissue. Previous result have demonstrated that Col6 knockout plays a crucial protective role after myocardial infarction through the inhibition of the myocardial infarction area, apoptosis, cardiac remodelling and fibrosis, leading to improved cardiac function. 11
The inflammatory response is essential for cardiac repair after myocardial I/RI and is involved in postinfarction remodelling of the myocardium and heart failure. Several proinflammatory cytokines, such as interleukin (IL)-1β, IL-6 and tumour necrosis factor (TNF)-α, are significantly overexpressed after myocardial infarction. 12 The overexpression of proinflammatory cytokines induces the synthesis of endothelial cell adhesion molecules, regulates adhesion interactions, and ultimately leads to inflammatory cell infiltration in the infarcted area. 13
Recent studies have confirmed that genetic or immunotherapeutic agents can ameliorate cardiac fibrosis and improve heart function.14-16 Moreover, certain pharmacological agents have been demonstrated to attenuate or reverse fibrosis, consequently partially restoring ventricular contractility and slowing the progression of myocardial failure. 17 Antifibrotic treatment is a chronic process. Artesunate, a traditional Chinese medicine, targets myeloid differentiation factor 2 (MD2) to alleviate myocardial fibrosis. However, its long-term effect still requires more clinical research for evaluation. 18 Currently, the FDA has not approved any drugs specifically targeting anti-myocardial fibrosis.19,20 Regarding the idiopathic anti-pulmonary fibrosis drugs pirfenidone and nintedanib, their performance in treating myocardial fibrosis has been disappointing. 21 They only provide moderately reduced myocardial fibrosis without improving cardiac function. 22 Therefore, there is an unmet need for clinical treatment of fibrosis in patients with myocardial I/RI.
Borojó is a fruit belonging to the Rubiaceae family, is produced mainly in the tropical rainforests of Colombia, and is a major source of iridoid glycosides. It has been reported that Borojó has anti-inflammatory, wound healing, and immunomodulatory properties. 23 We identified the active component of Borojó as 4,8-dicarboxy-8,9-cycloiridoid-1-glycoside (BIG) by nuclear magnetic resonance (NMR) and infrared spectroscopy, with the structure of C16 H20 O11 and shown in Figure 6F.24,25 Moreover, our research confirmed that BIG alleviates osteoarthritis by inhibiting proinflammatory cytokines and apoptosis. 26 However, it is worth studying whether BIG has a protective effect on the myocardium subjected to I/RI.
The main purpose of this study was to explore the mechanism by which BIG treatment protects the heart from myocardial I/RI: (1) whether BIG inhibits inflammation and apoptosis; (2) whether BIG inhibits myocardial fibrosis; and (3) whether BIG improves cardiac function.
Materials and Methods
Experimental Animals and Drugs
All the experimental animals were healthy adult male Sprague‒Dawley mice (weighing 200–250 g) purchased from Beijing Gene Line Bioscience Co. Ltd. (China). All the experimental procedures in this study were conducted in accordance with the ethical guidelines of Beijing Gene Line Bioscience Co., Ltd., and were approved by the Animal Ethics Committee of Beijing Gene Line Bioscience Co., Ltd. (Approval No. JLHK-20230110-01), and followed ARRIVE guidelines 2.0. The exclusion criteria for experimental mice were as follows: body weight deviation from the mean value ±20%; electrocardiogram detection showed arrhythmia and abnormal ST-T segment before the experiment; deviation in the ligation position of the left anterior descending coronary artery; reperfusion operation errors. This study was conducted between May 2023 and June 2025.
The drug BIG in this study was purchased from Guangzhou Baolukang Biotechnology Co., Ltd., China, with batch number 20210715. It is a white powder and readily soluble in pure water. The molecular structure was identified as C16H20O11 by High performance liquid chromatography-mass spectrometry (HPLC-MS) and NMR, and exact mass: 388.10, purity: 98%.
Mice Myocardial I/RI Model
Experimental mice were anaesthetized by intraperitoneal injection of pentobarbital sodium (60 mg kg-1), and after a few minutes, the tension in the limbs disappeared, confirming complete anaesthesia. The experimental mice were intubated and connected to a ventilator (Kent Scientific, SomnoSuite) for mechanical ventilation (inhaling 80% oxygen and 2% carbon dioxide, with a respiratory rate of 100 to 120 breaths min-1 and a tidal volume of 150 to 200 µl), and maintained the end-expiratory carbon dioxide (EtCO2) within the physiological range of 35-45 mmHg by adjusting ventilation parameters.
The experimental mice were fixed in the supine position on the operating plate, and the hair in the precordial area was removed and disinfected. Afterwards, a 2-cm incision was made between the 2,3 intercostal space, the thoracic cavity was opened, and the LAD was carefully exposed. A 5-0 surgical suture was passed under the LAD, and the suture was tightened to induce myocardial ischaemia. Successful LAD ligation was monitored by electrocardiogram, which revealed abnormal ST-segment elevation and the corresponding area of the left ventricle turning pallor in colour. The sham group underwent only thoracotomy without LAD ligation. After 45 minutes, the suture was loosened to achieve myocardial I/RI. The intercostal spaces were closed, and the muscles and skin were sutured. After the mice resumed spontaneous breathing, the tracheal intubation was removed, and they were allowed to breathe 100% oxygen until they began to move.
Experimental Design
In this study, pretreatment and therapeutic protocols were designed to explore the cardioprotective effect of BIG on myocardial I/RI. SD mice were randomly divided into 3 groups according to random numbers generated using the RAND function in Excel: the Sham, I/RI, and BIG groups, with 10 mice in each group. The BIG solution (98% original purity) was dissolved in sterilised saline to a concentration of 200 µg ml-1. The mice in the BIG group were intraperitoneally injected with 400 µg kg-1 BIG solution as a pretreatment one day before myocardial I/RI surgery. Afterwards, myocardial I/RI surgery was performed on the second day, and 400 µg kg-1 BIG solution was intraperitoneally injected daily from Days 2 to 7 as a therapeutic method.
We used the PI3K inhibitor LY294002 (50 μM; MedChemExpress, Shanghai) to explore whether the potential mechanism through which BIG protects against myocardial I/RI is related to the activation of the PI3K pathway. SD mice were randomly divided into 4 groups according to random numbers generated using the RAND function in Excel: the Sham, BIG + I/RI, BIG + Sham, and LY294002 + BIG + I/RI groups, with 10 mice in each group.
The mice in the LY294002 + BIG + I/RI group were intraperitoneally injected with LY294002 at a dosage of 40 mg kg-1 30 minutes before LAD ligation. Additionally, the researchers remained blinded to all procedures, including echocardiography, histological quantification, and data analysis, during all the experiments.
Cell Culture and Oxygen-Glucose Deprivation (OGD)
H9C2 (2-1) (Procell, CL-0089) cells were cultured in DMEM (Gibco, C11965500BT) supplemented with 10% foetal bovine serum (Gibco, A3160802) and 1% penicillin‒streptomycin in an incubator with 5% CO2 at 37 °C. An in vitro model of myocardial I/RI was established by using OGD. H9C2 cells were cultured in glucose‒free DMEM supplemented with 1% O2, 5% CO2, and 94% N2 at 37 °C for 4 hours. Afterwards, the medium was changed to normal DMEM, and the cells were reoxygenated for another 12 hours under normoxic conditions. We pretreated H9C2 cells with BIG at a concentration of 50 µg/ml 1 hours before OGD. The activity of H9C2 cells was detected using a CCK-8 (REMARKABLE, B1099) in accordance with the operating guidelines.
Determination of Myocardial Infarct Size
Evans blue and 2,3,5-triphenyl tetrazolium chloride (TTC) double staining was performed to determine the myocardial infarction size. The experimental animals were anaesthetized again on the 8th day after myocardial reperfusion. A 2% Evans blue (Aladdin, E104208) solution (dissolved in PBS) was rapidly injected into the left ventricle. Half an hour after injection, the LAD was ligated, and the animals were sacrificed. Heart tissues were removed and frozen in a -20 °C freezer for 1 hour. Heart tissue was cut evenly into 2-mm-thick slices. Heart slices were immersed in a 2% TTC (MP, 0219989225) solution that was soluble in PBS (pH 7.4) and incubated in the dark at 37 °C for 30 minutes. After they were rinsed with PBS, the cells were fixed with 4% paraformaldehyde. The red area represents the living myocardium, the white area represents the infarcted area, and the normal myocardium appears blue. Each heart was cut into five consecutive slices, and image analysis was conducted in a double-blind manner. Two researchers independently calculated the average value to avoid subjective bias. The sum of the left ventricular area, the area at risk (AAR), and the infarction area for each slice was calculated. The infarction area was calculated as a percentage of the AAR, and the AAR was calculated as a percentage of the LV.
Echocardiography for the Assessment of Cardiac Function
The experimental animals were anaesthetized again on the 8th day after myocardial reperfusion. Echocardiographic images (MYLAB™ X5 VET) were used to assess the cardiac structure and function of the experimental animals. After the target images were obtained, a two-dimensional image of the left ventricle was acquired through the long-axis view at the right parasternal position. The probe angle was adjusted to the short-axis view at the level of the papillary muscles to ensure that the section was nearly circular for accurate measurement of the internal diameter of the left ventricle. M-mode ultrasound requires the selection of a sampling line on the two-dimensional image to generate a curve of cardiac structure movement, precisely capturing the images at the end of diastole (when the ventricle is maximally dilated) and the end of systole (when the ventricle is maximally contracted). The left ventricular end-diastolic diameter (LVIDd) and left ventricular end-systolic diameter (LVIDs) were measured and recorded at the papillary muscle level. FS and EF are based on the measurement parameters and can be calculated as follows: FS = (LVIDd - LVIDs)/LVIDd × 100%, reflecting the contractility of the LV; EF = [EDV (end-diastolic volume) - ESV (end-systolic volume)]/EDV × 100%.
TUNEL Staining
Cardiomyocyte apoptosis after cardiac I/RI was detected by a TUNEL kit (Qihai Biology, AT005-1). After the experimental mice were anaesthetized, their hearts were perfused with 4% paraformaldehyde and cold PBS solution. Afterwards, the hearts were cut into 20-µm-thick slices and embedded in paraffin at 4 °C overnight. Paraffin-embedded heart slices first needed to be dewaxed and hydrated. Afterwards, 4% paraformaldehyde was added to the slices, which were subsequently fixed at room temperature for 30 minutes. The fixed slices were added to PBS containing 0.1% Triton X-100 and incubated on ice for 2 minutes to increase the permeability of the cell membrane. After pretreatment, proteinase K treatment was performed at 37 °C for 15 minutes to expose the DNA break ends, followed by washing with PBS. The reaction solution containing TdT enzyme and fluorescently labelled dUTP was dropped onto the slices and incubated in a 37 °C wet box in the dark for 1 hour. The slices were then washed with PBS three times to remove the unbound fluorescent markers. Then, anti-fluorescence quenching mounting medium was added for sealing, and the stained slices were photographed with a fluorescence microscope (Nikon Ci-S). Under the microscope, the cell nuclei appeared blue, and the apoptotic cells appeared green. The TUNEL‒positive cell ratio was calculated as follows: green/blue × 100%.
Haematoxylin–Eosin Staining
The experimental animals were anaesthetized and sacrificed, and their hearts were rinsed with cold PBS, fixed with 4% paraformaldehyde and embedded in paraffin. In accordance with the manufacturer’s standard protocol, haematoxylin (Sangon Biotech Co., Ltd., Shanghai, 607317-0100)–eosin staining (Sangon Biotech Co., Ltd., Shanghai, E607321-0100) was performed, and histological changes in the myocardium were examined under an optical microscope (Nikon DS-U3). According to previous studies, myocardial injury includes interstitial oedema, changes in myocardial fibrosis, and the degree of subendocardial haemorrhage. 27
Immunofluorescence Staining
After the experimental animals were anaesthetized, their hearts were extracted and cut into 5-mm-thick frozen slices. Afterwards, the slices were washed with PBS. The membranes were disrupted with 0.3% Triton X-100 for 10 minutes and then were washed with PBS. PBS was removed, and sample blocking solution was added. The slices were incubated in a wet box at room temperature for 2 hours. The blocking solution was removed, and the myocardial cells were treated with primary antibodies against TNNI3 (1:50) (Bioss, BD-PE0227), Col6a3 (1:100) (Bioss, bs-0553R), and TGF-β (1:200) (Affinity, AF1027); the myocardial fibroblasts were then treated with primary antibodies against vimentin (1:100) (Bioss, BD-PE1985), Col6a3 (1:100), and TGF-β (1:200). The slices were incubated in a wet box at 4 °C for 12 hours. Next, the slices were washed with PBS to remove the primary antibodies. Goat anti-mouse IgG H&L (Alexa Fluor 594) and goat anti-rabbit IgG H&L (Alexa Fluor 488) secondary antibodies were added at a ratio of 1:200 in the dark and incubated at 37 °C for 2 hours. Nuclear staining was performed with DAPI for 10 minutes. The slices were mounted with anti-fluorescence quenching mounting medium. Images were taken at 200x magnification (Nikon DS-U3). TGF-β and Col6a3 expression is shown in red; TNNI3 and vimentin expression is shown in green; and DAPI staining is shown in blue.
Western Blotting
After the experimental animals were sacrificed, the tissues from the ischaemic areas were extracted and cut into small pieces. A BCA kit (Solarbio, PC0020) was used to determine the protein concentration. Protein separation was performed by electrophoresis on a 10% SDS (Sinopharm, 30166428) gel, after which the proteins were transferred to PVDF (Millipore, IPVH00010) membranes. The PVDF membranes were blocked with TBST containing 5% milk. Primary antibodies against Col6a3 (1:1000), TGF‒β (1:1000), MMP-2 (1:1000), MMP-9 (1:1000), TNF‒α (1:500), IL-1β (1:1000), IL-6 (1:1000), BCL-2 (1:1000), BAX (1:2000), cleaved-caspase 3 (1:1000), p-PI3K (1:1000), PI3K (1:1000), p-AKT (1:1000), AKT (1:3000), p70 S6k (1:1000), p-SMAD3(1:1000), SMAD3(1:2000) and GAPDH (1:5000) were diluted with TBST containing 2% BSA overnight. The PVDF membrane was then incubated with a secondary antibody (Bioss, bs-0295G-HRP). The membranes were exposed using an automatic chemiluminescence image analysis system (Tanon, 5200), and the grey values of the exposure results were analysed using ImageJ software, and the target protein levels were standardized according to the GAPDH loading control.
Statistical Analysis
IBM SPSS Statistics 27.0 software was used for all the statistical analyses. The data are presented as the mean ± SEM, with at least six independent experiments in each group. The dose-response curve was plotted using GraphPad Prism 8.2 non-linear regression. One-way ANOVA with Tukey’s multiple comparisons test was used for multiple group comparisons. A P value less than 0.05 was considered to indicate statistical significance.
Results
BIG can Enhance the Activity of H9C2 Cells in Vitro
First, the protective effect of BIG on cardiomyocytes was verified through the in vitro culture of H9C2 cells. H9C2 cells were pretreated with six different concentrations of BIG (0, 10, 20, 50, 100, and 200 µg/ml) for 1 hour, followed by OGD for 4 hours, and then cocultured with BIG and H9C2 cells. After 48 hours, a CCK-8 assay was used to evaluate the viability of the H9C2 cells. The mean effective concentration (EC50) of BIG was determined using nonlinear regression (curve fit) in GraphPad Prism 8.2. The results demonstrated that the EC50 was 50.32 µg/ml (95% CI: 37.18‒93.88), and the dose‒response curve was plotted and is shown in Figure 1A. Therefore, in subsequent experiments, H9C2 cells were treated with 50 µg/ml BIG in vitro. BIG protected H9C2 cells from OGD‒induced injury via the inhibition of inflammation and apoptosis.
BIG Protects H9C2 Cells From OGD-Induced Injury Through the Inhibition of Inflammatory Responses, Apoptosis, and Extracellular Matrix Secretion
To investigate the mechanism by which BIG protects H9C2 cells from OGD‒induced injury, we treated H9C2 cells with 50 µg/ml BIG under controlled conditions. The H&E results revealed that in the OGD group, some cells exhibited swelling, and fine red granular substances were detected within the cytoplasm under light microscopy. Additionally, some cells displayed oedema, whereas others appeared loose with vacuolation. However, the extent of swelling in OGD‒induced cells pretreated with BIG was significantly attenuated. The TUNEL results also confirmed that the apoptosis rate in the OGD group was significantly greater than that in the control group. However, BIG attenuated OGD‒induced apoptosis (Figure 1B and E).
Matrix metalloproteinases (MMPs) play crucial roles in preserving cardiac function following I/RI. The overexpression of MMPs results in the degradation of the extracellular matrix (ECM), which consequently impairs cardiac contractility. 28 Proinflammatory cytokines, including TNF‒α and IL-1β, have been shown to activate MMPs.29-31 The WB results indicated that in the OGD‒induced H9C2 cell line (Figure 1L), the expression of the gelatinases MMP2 and MMP9 (Figure 1F and G) and the expression of proinflammatory factors, including TNF‒α, IL-1β, and IL-6, were significantly increased in the OGD group compared with the control group (Figure 1H, I and J). Notably, treatment with BIG effectively reversed these effects.
In an in vitro myocardial I/RI model, the level of anti-apoptotic protein BCL-2 significantly decreased, 32 whereas in surviving cells, the levels of the autophagy protein BAX and the pro-apoptotic protein c-caspase-3 were decreased. 33 Our results further substantiate that BIG upregulates BCL-2 expression while downregulating BAX and c-caspase-3 expression, thus mitigating OGD‒induced apoptosis in H9C2 cells (Figure 1K, C and D).
BIG Alleviates Myocardial I/RI and Cardiac Dysfunction
Considering that BIG has been demonstrated to effectively enhance the activity of the H9C2 cell lines in an OGD model in vitro, we hypothesized that BIG may exert a protective effect on cardiac function against I/RI. The echocardiography results revealed significant increases in the LVIDs and LVIDd in the I/RI group compared with those in the Sham group. However, BIG alleviated only the increase in LVIDs; the increase in LVIDd was not alleviated compared with that in the I/RI group. Furthermore, no difference in LVIDs or LVIDd was observed between the BIG and Sham groups (Figure 2E and F). BIG effectively alleviated cardiac dysfunction and inhibited cardiomyocyte apoptosis following I/RI.
Next, we proceeded to assess cardiac function in detail. Echocardiography confirmed that BIG significantly increased the ejection fraction and fractional shortening (Figure 2B and C), indicating that BIG effectively protected cardiac function against I/RI. Given that the infarct size of the myocardium is closely associated with cardiac function, investigating whether BIG can reduce the myocardial infarction area following I/RI is essential. The TTC results demonstrated that BIG significantly decreased the myocardial infarction area and area at risk (AAR) compared with those in the I/RI group (Figure 2G and H). Next, we employed haematoxylin and eosin (H&E) staining to verify the influence of BIG on the pathological progression of I/RI. In the I/RI group, cardiac injury was characterized by haemorrhage, oedema, proliferation of fibroblasts and fibrocytes, and infiltration of inflammatory cells and macrophages. However, BIG significantly mitigated the tissue oedema, fibrous tissue proliferation, and inflammatory cell infiltration observed at the site of left ventricular injury (Figure 2J).
Furthermore, we used a TUNEL assay to analyse the effect of BIG on cardiomyocyte apoptosis. TUNEL staining clearly demonstrated that BIG markedly attenuated cardiomyocyte apoptosis following I/RI (Figure 2I).
BIG Alleviates I/R-Induced Inflammatory and Apoptotic Responses in Vivo
Because inflammation is closely associated with adverse cardiac events following ischaemia/reperfusion injury (I/RI), 34 we subsequently investigated whether BIG could suppress the inflammatory response induced by I/RI.
The WB results showed that the protein levels of proinflammatory factors, such as TNF‒α, IL-1β, and IL-6, as well as MMP2 and MMP9 (Figure 3A-E), were markedly lower in mice treated with BIG than in those in the I/RI group. These findings suggest that BIG has the potential to suppress the inflammatory response induced by I/RI. Cardiomyocyte apoptosis plays a critical role in the progression of I/RI.
35
The inhibition of cardiomyocyte apoptosis is associated with an increased Bcl-2/BAX ratio, which promotes the survival of myocardial cells at the infarct border zone.
36
Therefore, the Bcl-2/BAX ratio is usually used as an indicator to predict the level of apoptosis. Our WB results also confirmed that BIG upregulates BCL-2 (Figure 3F) expression while downregulating BAX (Figure 3H) expression, consequently increasing the Bcl-2/BAX ratio. This alteration is closely associated with the suppression of the expression of the apoptosis-related protein cleaved‒caspase 3(Figure 3I). TUNEL staining further confirmed that BIG notably reduced the degree of apoptotic death of cardiomyocytes (Figure 3K). BIG improves cardiac function after I/RI by inhibiting the inflammatory response and myofibroblast fibrosis.
BIG Prevents Cardiac Fibrosis in I/RI Model Mice
During the process of myocardial I/RI repair, myocardial fibroblasts are activated, leading to extensive myocardial fibrosis, which is characterized by excessive deposition of collagen and other extracellular matrix (ECM) proteins in the myocardium. This pathological remodelling eventually results in cardiac dysfunction. 37 Repair of fibrosis involves the replacement of myocardial cells with collagen following acute myocardial I/RI, a process primarily mediated by fibroblasts and myofibroblasts. 4 Transient repair of fibrosis plays an essential role in maintaining cardiac structure. However, persistent and excessive fibrosis results in aberrant ECM deposition and cardiac remodelling, ultimately leading to heart failure. Vimentin and troponin I type 3 (TNNI3) are considered key markers of cardiac fibroblasts and cardiomyocytes, respectively. 3 In the initial phase of myocardial I/RI, fibroblasts or myofibroblasts are activated by various cytokines, including TGF‒β, resulting in an increase in myocardial fibrosis during the subsequent reparative period. 38 Our immunofluorescence results demonstrated that the number of TGF‒β‒ and Col6a3-positive cells among cardiac fibroblasts was significantly greater in the I/RI group than in the Sham group, whereas BIG effectively reversed these effects (Figure 3L and M). However, the numbers of TGF‒β‒ and Col6a3-positive cardiomyocytes did not significantly differ between the I/RI group and the Sham group (Figure 3P and Q).
Taken together, these results illustrate that BIG can alleviate myocardial fibrosis after I/RI and thus improve cardiac function.
Cellular Atlas of Myocardial Infarction
In this previous study, a total of 31 samples were collected, including 4 normal nontransplanted donor hearts as the control group, 12 infarcted myocardial tissue samples, 3 necrotic border zone tissue samples, 6 remote zone tissue samples, and 6 fibrotic zone tissue samples.
39
Thus, a map of human heart cell types was constructed. Single-nucleus RNA sequencing (snRNA-seq) and cluster analysis revealed 10 major cardiac cell types (Figure 4A). We utilized a differential expression analysis tool based on the data (https://cellxgene.cziscience.com) to identify 132 significantly differentially expressed genes (67 upregulated and 65 downregulated, log fold change above ±1, P < 0.05) in fibroblasts from myocardial infarction tissues compared with those from normal cardiomyocytes (Figure 4C); moreover, compared with these genes, the expression of Col6a3 was the highest (Figure 4B). Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis revealed that the differentially expressed genes were enriched in the PI3K/AKT signalling pathway (Figure 4D). Changes in the multiomic map after myocardial infarction.
Next, we extracted tissue from the myocardial infarction area, and WB also confirmed that the protein levels of TGF‒β and Col6a3 were greater on the 8th day after myocardial I/RI in the I/RI group than in the sham group, whereas BIG significantly inhibited the expressions of TGF‒β and Col6a3 (Figure 4E).
BIG Improved Cardiac Function Following I/RI by Enhancing the Phosphorylation of PI3K/AKT
BIG promotes cardiac function after myocardial I/RI. To explore whether this occurs mechanologically through the PI3K/AKT signaling pathway, the PI3K-specific inhibitor LY294002 was used to verify this hypothesis. LY294002 was intraperitoneally injected at a dosage of 40 mg/kg 30 minutes before myocardial ischaemia. Transthoracic echocardiography revealed that BIG did not promote cardiac function in the sham group; BIG improved cardiac function after I/RI, and there was no difference compared with that in the sham group (Figure 5A). However, after pretreatment with the PI3K inhibitor LY294002, EF and FS were significantly reduced, and the cardioprotective effect of BIG was abolished (Figure 5B and C). Similarly, LY294002 increased LVIDd and LVIDs (Figure 5D and E), indicating that LY294002 impaired the contractile and diastolic functions of the heart. The WB results confirmed that BIG upregulated the expression of phosphorylated PI3K and AKT, and accordingly, the level of p70 S6 kinase, a downstream target of the PI3K/AKT signaling pathway, was also upregulated (Figure 5H). However, LY294002 prevented this protective effect of BIG (Figure 5J). Inhibition of PI3K abolished the BIG-induced cardioprotective effect following myocardial I/RI.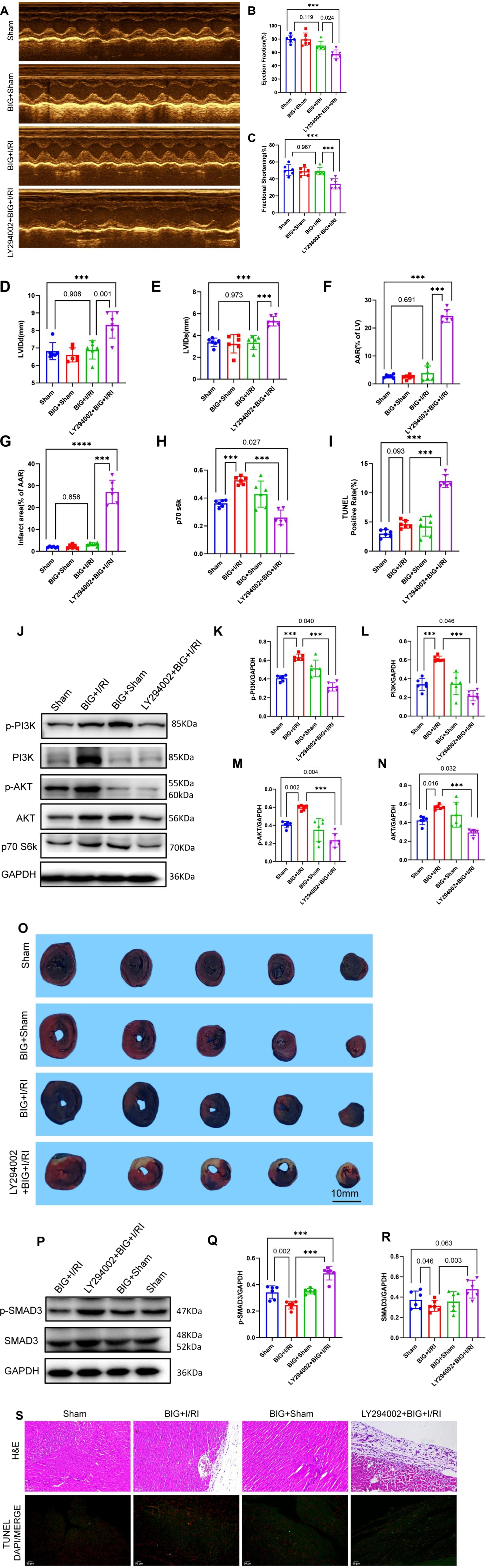
Smad3 (mothers against decapentaplegic homolog 3), which is downstream of TGF‒β, is a crucial driver of myocardial fibrosis. It can directly bind to the promoters of collagen (Col1a1, and Col3a1) to promote ECM synthesis. The TGF‒β/SMAD3 signaling pathway upregulates fibronectin, collagen, α‒SMA, and connective tissue growth factor, and is widely involved in myocardial fibrosis and cardiac remodeling. The progression of cardiac fibrosis after myocardial infarction was alleviated by inhibiting the TGF‒β/SMAD3 signaling. 10 Our results confirmed that BIG inhibits the expression of p-Smad3 and suppresses myocardial fibrosis after myocardial I/RI. However, LY294002 eliminated this protective effect of BIG (Figure 5Q).
TTC staining (Figure 5O) revealed that BIG did not increase the AAR (Figure 5F) or infarct size (Figure 5G) following I/RI compared with those in the Sham group. However, LY294002 reversed the protective effect of BIG, the AAR and infarct area significantly increased, and cardiac function was severely impaired. TUNEL staining also confirmed that LY294002 increased cardiomyocyte apoptosis (Figure 5I). The H&E staining results revealed that in the sham group, the normal myocardial fibres were irregular, short, and cylindrical in shape, with branches interconnected into a network. The nuclei were normal, with a few binucleated, oval in shape, and located in the centre of the cells. The I/RI myocardium treated with BIG shows partial absence of the myocardial fibrous basement membrane and destruction of the plasma membrane, and the cellular structure has not yet been destroyed. The microvessels dilate and swell because of blockage, and damaging changes can be observed in the endothelial cells. However, after pretreatment with LY294002, the basal membrane of the myocardial fibres was completely absent, the plasma membrane was damaged, severe contraction bands appeared in the cell structure, and the muscle filaments were broken. The microvessels dilate and swell because of blockage, and damaging changes can be observed in the endothelial cells (Figure 5S).
AKT Inhibitors Abolished the Protective Effect of BIG Against OGD-Induced Injury in H9C2 Cells
Previous studies have shown that the downstream kinase AKT of PI3K can transduce enzymes that protect the myocardium from I/RI.
40
Given that AKT plays a key role in the survival of cardiomyocytes,41,42 we further utilized an AKT inhibitor (Aladdin, 32387-96-5) to verify the mechanism by which BIG protects H9C2 cells from OGD-induced injury. The TUNEL results confirmed that BIG could significantly inhibit the apoptosis of H9C2 cells induced by OGD. However, the AKT inhibitor counteracted this protective effect of BIG on H9C2 cells (Figure 6D). WB also revealed that BIG promoted high levels of phosphorylated AKT and PI3K in H9C2 cells subjected to OGD, whereas the AKT inhibitor reversed the upregulation of phosphorylated AKT and PI3K induced by BIG (Figure 6B and C). These findings clarified that AKT was indispensable for the ability of BIG to protect H9C2 cells from OGD injury. The H&E staining results revealed that OGD caused regional degeneration and necrosis of H9C2 cells, with disordered arrangement, myofilament rupture and dissolution, unclear striations, nuclear pyknosis, and perinuclear vacuoles, accompanied by inflammatory cell infiltration. In H9C2 cells pretreated with BIG, small areas of degeneration were still observed, but no obvious necrosis was observed, the arrangement of H9C2 cells was slightly irregular, and nuclear pyknosis was visible. However, pretreatment with an AKT inhibitor reversed the protective effect of BIG on OGD‒induced injury in H9C2 cells. The AKT inhibitor caused the degeneration and necrosis of H9C2 cells, with a slightly disordered arrangement, abnormal cell morphology, nuclear pyknosis, and inflammatory cell infiltration (Figure 6E). The inhibition of AKT abolished the protective effect of BIG on H9C2 cells subjected to OGD.
Discussion
The most crucial finding of this study is that BIG can significantly reduce the myocardial infarction area after cardiac I/RI and improve cardiac function. Both our in vivo and in vitro studies confirmed that BIG protects cardiomyocytes by significantly inhibiting the expression of proinflammatory factors, ECM components and apoptosis-related proteins. BIG inhibited the expression of Col6a3 and TGF‒β in cardiac fibroblasts but not in cardiomyocytes, thus reducing fibrosis in the myocardial infarction area after I/RI and protecting cardiac function (Figure 3N and O). Additionally, we verified that BIG protects cardiac function after I/RI by activating the PI3K/AKT signalling pathway. In vivo, the administration of the PI3K inhibitor LY294002 to inhibit PI3K abolished the protective effect of BIG on the heart against I/RI. Similarly, in vitro, the administration of an AKT inhibitor abolished the protective effect of BIG against OGD in H9C2 cells.
Repairative cardiac fibroblast activation peaks approximately one week after myocardial I/RI and can persist for years, ultimately leading to the formation of scars composed mainly of cross-linked collagen, causing left ventricular remodelling and reducing cardiac function. 43 How to reduce myocardial fibrosis after I/RI remains challenging. Many studies have provided promising perspectives. For instance, targeting fibroblast activation protein (FAP) in myofibroblasts, a vaccine against FAP, and chimeric antigen receptor (CAR) targeting FAP can improve cardiac function after I/RI.15,16,44 However, the cost-effectiveness of these immunotherapies remains a problem. TGF‒β plays a critical role in the repair process after myocardial infarction. Degradation of TGF‒β or the TGF‒β‒neutralizing antibody can markedly ameliorate cardiac fibrosis, left ventricular hypertrophy and systolic function.10,45 To our knowledge, previous studies have not investigated whether BIG improves cardiac function after myocardial I/RI and its related mechanisms. With the increase in the ageing population worldwide, an increasing number of geriatric patients are suffering from myocardial ischaemic diseases, necessitating more cost-effective treatment approaches to address cardiac I/RI. Our research used BIG as a treatment to protect the myocardium of mice from I/RI, providing a potential therapeutic strategy for patients with acute myocardial infarction in the future. Our results confirmed that Col6a3 and TGF‒β were highly expressed in the myocardial tissue of the infarct area on Day 8 after myocardial I/RI; however, BIG effectively inhibited the expression of Col6a3 and TGF‒β (Figure 4E). The authors further divided human cardiac fibroblasts into four subpopulations, Fib1- 4. Among them, Fib2 (marked by POSTN, COL1A1 and FN1) contains terminally differentiated myofibroblasts and expresses most ECM‒related genes. The Fib2 (myofibroblasts) is significantly expressed in myocardial ischaemic areas and increases ECM deposition. 39 Similarly, our results confirmed that BIG inhibited only the expression of Col6a3 and TGF‒β in cardiac fibroblasts (Figure 3N) but had no effect on their expression in cardiomyocytes (Figure 3O). However, notably, we verified that BIG activates PI3K/AKT, thereby inhibiting the expression of Col6a3 and TGF‒β. It is necessary to further verify whether BIG reduces the expression of Col6a3 and TGF‒β by using the PI3K inhibitor LY294002 or an AKT inhibitor to decrease the expression of PI3K or AKT. These findings directly confirm that the expression of Col6a3 and TGF‒β induced by BIG occurs through the PI3K/AKT pathway. Cardiac fibroblasts are tissue-specific fibroblasts, whereas primary cardiac fibroblasts are initially cultured cells directly isolated from cardiac tissue. BIG inhibits the expression of Col6a3 and TGF‒β in cardiac tissue fibroblasts, thereby suppressing myocardial fibrosis after I/RI. This requires further verification in primary cardiac fibroblasts.
In this study, we administered an intraperitoneal injection of BIG on the basis of the following considerations. As a small-molecule water-soluble drug, BIG has a stable increase in blood concentration after intraperitoneal injection, and its bioavailability reaches 90%. Sterilising saline as its carrier does not affect its efficacy. However, the blood concentration of BIG increases slowly when BIG is orally administered, and its bioavailability is only 70%, which reduces the efficacy of the drug. Although BIG reaches its peak blood concentration quickly and has 100% bioavailability when administered intravenously, this procedure is difficult.
The proinflammatory cytokines released by neutrophils are critical mediators of inflammation and myocardial remodelling after myocardial infarction. Regulating the inflammatory response and repair process may be an effective strategy for treating myocardial ischaemia.9,46 During the repair process of cardiac I/RI, the rational regulation of ECM deposition and cross-linking proteins is conducive to preventing dilated remodeling while avoiding excessive fibrosis and diastolic dysfunction. Conversely, the excessive activation of proteolysis induced by MMPs may promote left ventricular dilated remodeling, thus reducing systolic function. How to balance matrix synthesis and matrix degradation is crucial for repair after myocardial I/RI. Our findings confirmed both in vitro (Figure 1F, G, H, I and J) and in vivo (Figure 3A, B, C, D and E) that BIG significantly decreased the levels of MMPs and the inflammatory response after myocardial I/RI. Erya Chen et al. 47 pretreated mice with the Toll-like receptor ligand polyinosinic-polycytidylic acid (poly(I:C)) to protect the heart against I/RI. These findings are consistent with our findings in that both BIG and poly(I:C) significantly improved EF and FS and reduced LVIDs. However, neither BIG nor poly(I:C) significantly reduced LVIDd (Figure 2E). These findings suggest that the therapeutic strategies involving BIG and poly(I:C) for inhibiting cardiac fibrosis still require extensive study. Re-evaluating cardiac function 3 to 6 months after myocardial I/RI is extremely important, as it can reduce the risk of long-term cardiovascular events. Our findings clarify that BIG can inhibit myocardial fibrosis in the short term, but the long-term improvement in cardiac function after myocardial I/RI requires further research.
Although inhibiting cardiac fibrosis is regarded as the most attractive therapeutic approach in the repair process of cardiac I/RI, several challenges need to be overcome in practical applications. First, cardiomyocytes lack effective regenerative capacity, and cardiac fibrosis is an activation of a repair program rather than a primary pathological process; thus, establishing a causal relationship between ECM deposition and cardiac dysfunction is difficult. Second, the excessive fibrotic response in the heart is a cause of heart dysfunction and heart failure, for which a wide range of biomarkers are needed for identification. Third, the activation of fibroblasts and the disruption of the ECM network after cardiac I/RI and the continuous application of drugs that inhibit fibroblast activation may pose risks. Finally, antifibrotic strategies after cardiac I/RI need to be confirmed in more clinical studies.
Apoptosis plays a key role in the pathogenesis of myocardial I/RI. Inhibiting apoptosis may reduce the degree of myocardial damage and prevent the injury caused by myocardial I/R. The BCL-2 protein family regulates cardiomyocyte apoptosis after cardiac I/R, increasing the synthesis of caspase-3 and BAX while inhibiting the expression of BCL-2. 48 Our findings also confirmed that BIG protected H9C2 cells from OGD‒induced injury in vitro by inhibiting apoptosis, as it suppressed the expression of c-caspase-3 and BAX while upregulating the expression of the antiapoptotic protein BCL-2 (Figure 1C, D and K). Similarly, BIG protected the heart from myocardial I/R-induced injury in vivo by inhibiting cardiomyocyte apoptosis (Figure 1H, I and J).
The repair process after myocardial ischaemia indicates that several signaling pathways are involved in the survival, apoptosis and autophagy of cardiomyocytes and fibroblasts. These signaling pathways include the NLRP3/caspase-1 inflammatory response, the regulation of myocardial fibrosis by TGF‒β/SMADs, and the regulation of angiogenesis by PI3K/Akt. 49 More importantly, as a key prosurvival pathway, the PI3K/Akt pathway plays a crucial role in the repair process of myocardial I/RI. Recently, several studies have confirmed that the PI3K/Akt signaling pathway plays important roles in inhibiting the inflammatory response, antiapoptotic activity and myocardial fibrosis after myocardial I/RI.47,50
Bioinformatics analysis revealed that the genes that were differentially expressed between the myocardial infarction area and normal myocardial tissue were enriched in the PI3K/AKT signaling pathway (Figure 4D). To explore the mechanism by which BIG protects the heart against myocardial I/R-induced injury, we first used the PI3K-specific inhibitor LY294002 to verify that the activation of PI3K by BIG is indispensable. Our results demonstrated that compared with the sham group, BIG treatment significantly increased the levels of phosphorylated PI3K and AKT in hearts subjected to I/RI. Additionally, p70 S6K, a downstream protein kinase of phosphorylated Akt, is associated with protection against myocardial I/RI, 51 and its expression level is also increased. However, LY294002 abolished the protective effect of BIG (Figure 5J). Echocardiography revealed that BIG protected the mice heart from I/RI, and there was no difference in cardiac function compared with that in the Sham group. However, LY294002 reversed the protective effect of BIG, with significant decreases in EF and FS (Figure 5A). BIG did not increase the infarction area or AAR after myocardial I/RI compared with those in the Sham group (Figure 5O); these results revealed the protective effect of BIG on cardiac function. Regrettably, LY294002 significantly increased the myocardial infarction area and AAR, which led to severe impairment of cardiac function (Figure 5F and G).
Admittedly, as a classic PI3K inhibitor, the off-target effect of LY294002 mainly stems from the non-specific inhibition of CK2 and DNA‒PK, and LY294002 may also affect the mTOR pathway at high concentrations. Optimising the drug concentration and incubation time of LY294002 is important for avoiding its off-target effects. LY294002 at concentrations ranging from 1–5 μM not only effectively inhibited the PI3K pathway but also reduced the off-target inhibition of CK2 and DNA‒PK. With respect to the dose, 47 we selected LY294002 at 40 mg kg-1 i.p. and maintained the incubation period at 7 days, mainly to avoid off-target effects of LY294002. Of course, the exact mechanism by which BIG activates PI3K still requires cross-verification through multiple orthogonal experiments. With respect to the PI3K catalytic subunit p110α, siRNAs were designed to knock down the expression of PI3K or overexpress PI3K, and the CK2 inhibitor CX-4945 or the DNA‒PK inhibitor Nu7441 alone were used to further verify that the protective effect of BIG against myocardial I/RI occurred through the PI3K pathway.
Previous studies have confirmed that overexpression of Akt can reduce the myocardial infarction area in rat hearts subjected to I/RI, 52 whereas transfection of Akt siRNA worsened cell death induced by OGD, indicating that Akt is a pro-survival protein in the myocardium. Activation of the PI3K/Akt signaling pathway can reduce apoptosis. 53 We used an AKT inhibitor to verify this hypothesis. Our findings confirmed that in vitro OGD inhibited the phosphorylation of PI3K and AKT in H9C2 cells but that BIG significantly increased the phosphorylation of PI3K and AKT. However, the AKT inhibitor suppressed the high levels of PI3K and AKT phosphorylation induced by OGD. Similarly, the AKT inhibitor reversed the increase in the levels of phosphorylated PI3K and AKT caused by BIG (Figure 6A). Similarly, BIG inhibited OGD‒induced apoptosis in H9C2 cells, and AKT inhibition reversed the protective effect of BIG on H9C2 apoptosis caused by OGD (Figure 6D). Therefore, our data indicated that the cardioprotective effect of BIG against myocardial I/RI was due to the activation of the PI3K/AKT signaling pathway.
Although our results demonstrated that BIG protects H9C2 cells from OGD injury in an AKT‒dependent manner in vitro, it is notable that H9C2 cells retain some key characteristics of cardiomyocytes and are a commonly used alternative model for in vitro research on the biological functions of cardiomyocytes. H9C2 cells can successfully replicate the characteristics of OGD, such as cardiomyocyte apoptosis, mitochondrial dysfunction, and oxidative stress activation. However, in some studies focusing on myocardial stunning, reperfusion arrhythmias, and other aspects directly related to myocardial contractility or electrical activity, primary cardiomyocytes are more advantageous.
This study clearly has several limitations. First, we verified only that BIG inhibited the expression of Col6a3 and TGF‒β in cardiac fibroblasts after I/RI and demonstrated its antifibrotic effect, but we did not further verify the antifibrotic effect of BIG by using Col6a3 and TGF‒β protein overexpression or knockdown methods in vivo and siRNA transfection in vitro. Of course, the genetic map of BIG altering myocardial I/RI requires further research. Second, the formation and remodelling of the infarct area in hearts subjected to I/RI is a long-term process. We detected only the infarct area and cardiac function on Day 8 after myocardial I/RI. The time window of the protective effect of BIG against I/RI in the heart needs further study. Third, this study verified only that BIG protects the heart against I/RI via the PI3K/AKT signaling pathway. The signalling pathway involved in the cardioprotective effect of BIG requires precise localisation through proteomic sequencing and KEGG analysis. Although apoptosis is inhibited in female mice through the oestrogen receptor-mediated PI3K/AKT pathway, the results of this study, which used only male mouse models, cannot be accurately extrapolated to female individuals, which may lead to poor efficacy or reduced safety of clinical treatment regimens in the female population. Finally, in the in vivo experiments section, we used only a single dose of 400 µg⋅kg-1 BIG, and the optimal dose of BIG should be determined through dose‒response experiments.
Conclusions
Our results indicate that BIG protects the heart against I/RI and reduces inflammatory and apoptotic responses and the expression of MMPs, which are regulated by the activation of the PI3K/AKT signalling pathway. These findings further confirm that the overexpression of Col6a3 and TGF‒β in cardiac fibroblasts after the heart is subjected to I/RI plays a critical role in regulating myocardial fibrosis. The results of the current study are conducive to understanding the relationship between BIG and downstream signalling molecules, highlighting the protective effect of BIG after myocardial I/RI and providing a scientific basis for its use as an effective therapeutic strategy. Our findings support the use of BIG as a potential therapeutic drug for cardiac I/RI.
Footnotes
Acknowledgements
Ethical Considerations
All the experimental procedures in this study were conducted in accordance with the ethical guidelines of Beijing Gene Line Bioscience Co., Ltd., and were approved by the Animal Ethics Committee of Beijing Gene Line Bioscience Co., Ltd. (Approval No. JLHK-20230110-01).
Author Contributions
ZPH and WBW conceived and designed the project, ZPH supervised this study; AJS established the myocardial I/RI model in experimental mice. CYY and FYZ conducted in vivo and in vitro experiments and collected data. AJS and WBW analyzed these data. WBW was responsible for all images and figures. JC and LCC were in charge of cardiac ultrasound operations and analyzed the data. AJS wrote the manuscript, and ZPH and WBW revised the final version. All authors read and approved the final manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by the Key Projects of Natural Science Research in Universities of Anhui Province (NO: 2024AH051150), Anqing Medical College Clinical Research Center (Anqing Municipal Hospital), Anqing, Anhui P.R China. AJS held the foundation.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
All data and materials of this study are available from both corresponding authors upon reasonable request.