
Abstract
Objectives
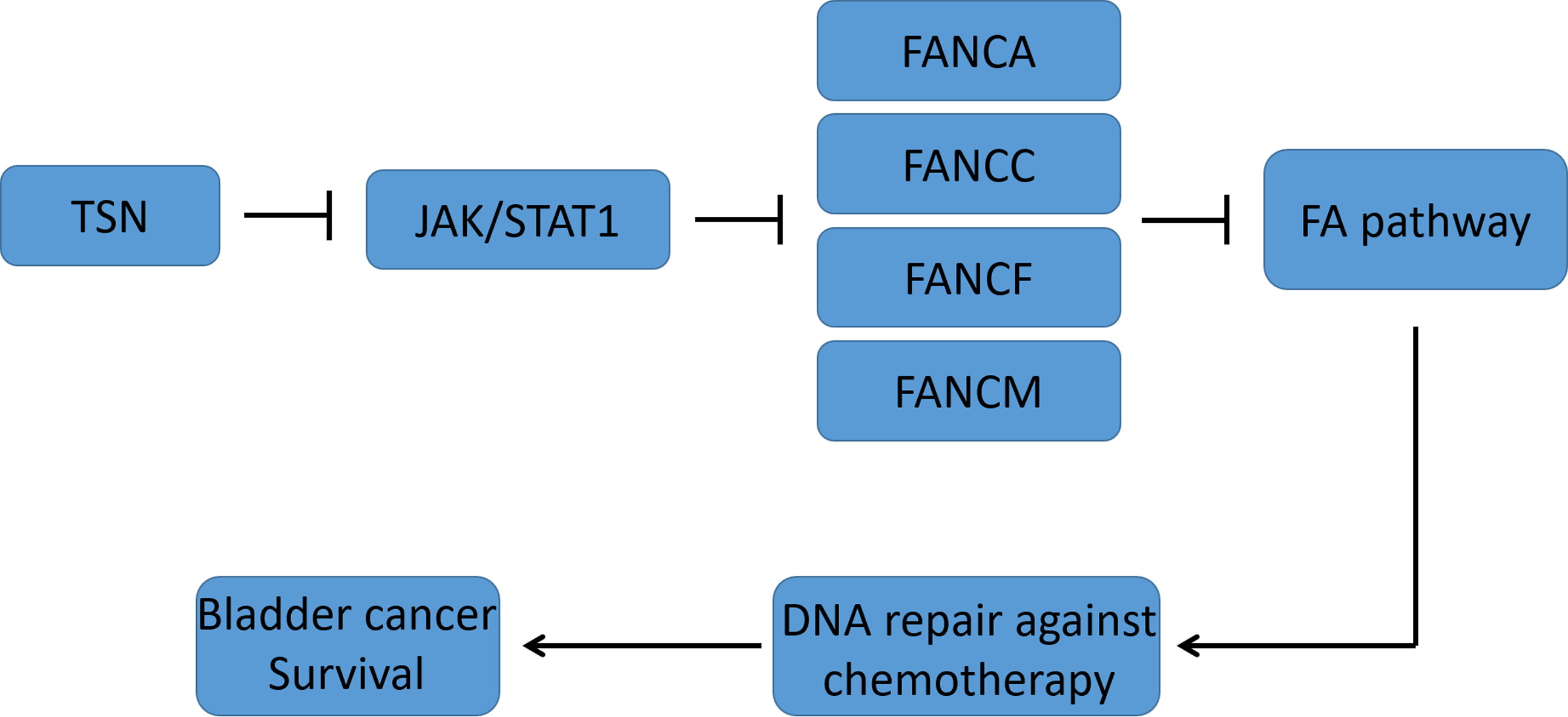
Resistance to platinum-based chemotherapy in bladder cancer is closely associated with activation of the Fanconi anemia (FA) DNA interstrand crosslink repair pathway. Identifying pharmacological inhibitors of FA signaling may improve therapeutic response. We investigated the effects of the natural compound toosendanin (TSN) on FA pathway regulation in this study.
Methods
Bladder cancer cell lines (T24, RT4, J82) were pretreated with TSN and exposed to ultraviolet C (UVC). FANCI/FANCD2 monoubiquitination, FANCD2 nuclear foci, FA core gene expression, and JAK/STAT1 signaling were quantified. A T24 xenograft model was used to validate FA pathway inhibition in vivo. Statistical analyses were performed using one-way ANOVA followed by Tukey’s post-hoc test (mean ± SD; n = 3 independent replicates; Shapiro–Wilk test for normality).
Results
TSN reduced UVC-induced 58% FANCI (P = 0.004, n = 3) and 77% FANCD2 monoubiquitination (P = 0.004, n = 3) in bladder cancer cells, and decreased FANCD2 foci-positive nuclei by 27% (P = 0.015, n = 3). Co-immunoprecipitation assays further revealed that TSN disrupted 56% interaction between the FANCI-FANCD2 complex and the key scaffold protein SLX4 (FANCP) (P = 0.002, n = 6). TSN down-regulated FA core genes (FANCA, FANCC, FANCF, FANCM) by 30 to 65% (all P < 0.05, n = 3) as well as decreased STAT1 phosphorylation by 45% (P = 0.013, n = 3) and the binding capacity of STAT1 on these FA genes’ promoter by 33% to 47% (all P < 0.05, n = 3). In xenograft tumors, TSN also reduced 70% FANCI (P = 0.007, n = 3) and 45% FANCD2 monoubiquitination (P = 0.011, n = 3) as well as expression of FANCA, FANCC, FANCF, FANCM by 26% to 38% (all P < 0.05, n = 3). TSN selectively sensitized bladder cancer cells to UVC-induced cytotoxicity (IC50 decreased 35%, P = 0.026, n = 3), without affecting the viability of human urothelial cell SV-HUC-1 cells or lung adenocarcinoma A549 cells (both P > 0.05, n = 3).
Conclusion
TSN inhibits FA DNA repair signaling in bladder cancer by suppressing JAK/STAT1-mediated FA core gene transcription, supporting its potential as a combinatorial agent to overcome cisplatin resistance.
Introduction
Bladder cancer is one of the most common malignancies of the urinary system, characterized by high recurrence rates and therapeutic resistance.1,2 Despite advances in surgery, chemotherapy, radiotherapy, and immunotherapy, treatment outcomes remain suboptimal for patients with advanced or muscle-invasive bladder cancer.3,4 Platinum-based chemotherapy, particularly cisplatin, is still a standard first-line regimen, yet many patients exhibit intrinsic or acquired resistance.5,6 Therefore, identifying molecular mechanisms underlying therapy resistance and developing new strategies to enhance treatment efficacy are urgently needed.7,8
The Fanconi anemia (FA) pathway is a DNA damage response mechanism that plays a critical role in maintaining genomic stability by repairing interstrand crosslinks, such as those induced by platinum-based agents.9-11 Core components of the FA pathway, including FANCI and FANCD2, undergo monoubiquitination in response to DNA damage, initiating repair through homologous recombination.12-14 In bladder cancer, activation of the FA signaling pathway has been associated with enhanced DNA repair capacity and resistance to DNA-damaging agents, suggesting that the FA pathway may serve as a potential therapeutic target to overcome chemoresistance. 15
Toosendanin (TSN), a triterpenoid extracted from the traditional Chinese medicinal plant Melia toosendan, has been reported to exhibit broad-spectrum anticancer activities. 16 Studies have demonstrated that TSN can inhibit tumor cell proliferation, induce apoptosis, and interfere with key oncogenic pathways across various cancer types.17-21 Emerging evidence suggests that TSN may also modulate DNA damage responses and sensitize tumor cells to chemotherapy.21-23 However, its precise mechanism of action remains to be fully elucidated. Investigating the interplay between TSN and DNA repair pathways, such as the FA signaling cascade, may reveal novel insights into its potential as a therapeutic adjuvant in bladder cancer.
Although TSN has been studied in several cancer types, no prior reports have investigated its therapeutic potential in bladder cancer. Furthermore, the relationship between TSN and DNA damage repair mechanisms - particularly the FA signaling pathway - remains poorly understood. Given that the FA pathway plays a critical role in mediating resistance to DNA-damaging agents such as cisplatin, it is essential to determine whether TSN can modulate this pathway and thereby influence therapeutic responses.
Therefore, in this study, we aimed to explore the functional role of TSN in bladder cancer and to elucidate the underlying molecular mechanisms by which it may regulate the FA signaling cascade. Specifically, we examined whether TSN treatment affected the activation of FA pathway components, such as FANCI and FANCD2 monoubiquitination, in response to genotoxic stress. Our findings provided novel insights into the interplay between natural compounds and DNA repair pathways and suggested that TSN might serve as a promising chemosensitizing agent for the treatment of bladder cancer.
Materials and Methods
Study Duration
This study was conducted from April 2023 to August 2024.
Cell Culture
Human urinary bladder carcinoma cell line T24 (ZQ0120), J82 (ZQ0348), human urinary bladder papilloma cell line RT4 (ZQ0350), SV40-immortalized human urothelial cell line 1 (SV-HUC-1, ZQ0345), human lung adenocarcinoma cell line A549 (ZQ0003) were obtained from Shanghai Zhong Qiao Xin Zhou Biotechnology Co., Ltd (China). All cell lines used in this study were authenticated by short tandem repeat (STR) profiling within 3 years prior to their use in the described experiments, and were routinely tested and confirmed to be free of mycoplasma contamination. T24 and J82 cells were cultured in RPMI-1640 medium (Gibco, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, USA), 100 U/mL penicillin, and 100 µg/mL streptomycin. RT4 cells were maintained in McCoy’s 5A medium (Gibco, USA) supplemented with 10% FBS and the same concentrations of antibiotics. All cells were incubated at 37°C in a humidified atmosphere with 5% CO2.
Ultraviolet radiation (UVC) exposure was performed using a UVP CL-1000 Ultraviolet Crosslinker (Analytik Jena, USA) equipped with 254 nm UVC lamps. Cells were first washed with phosphate-buffered saline (PBS), and the culture medium was replaced with a thin layer of PBS during irradiation to prevent light scattering. Cells were then exposed to a total UVC dose of 20 J/m2 over 20 s. After treatment, PBS was removed and replaced with fresh culture medium, and the cells were returned to the incubator for subsequent analyses.
Cells were incubated with the final concentrations of 100 nM Toosendanin (TSN, purity ≥98%, HY-N0263, MedChemExpress, China) for 24 h, 1 μM JAK/STAT1 inhibitor Ruxolitinib (HY-50856, MedChemExpress) for 24 h, 20 ng/mL JAK/STAT1 agonist interferon-gamma (IFN-γ, HY-P73252, MedChemExpress) for 1 h.
Western Blotting
1 × 106 cells were lysed in RIPA buffer (P0013 B, Beyotime, China) supplemented with protease and phosphatase inhibitors (P1045, Beyotime) on ice for 30 min. The lysates were centrifuged at 12 000 × g for 15 min at 4°C, and the supernatants were collected. Protein concentration was determined using the BCA protein assay kit (P0009, Beyotime). Equal amounts of 40 μg protein were separated by SDS-PAGE and transferred onto PVDF membranes (P0021S-1L, Beyotime). Membranes were blocked with 5% non-fat milk in TBST for 1 h at room temperature and incubated overnight at 4°C with primary antibodies against FANCI (20789-1-AP, Proteintech, China, 1: 2000), FANCD2 (NB100-316, Novus Biologicals, USA, 1: 2000), β-Actin (AC026, ABclonal, China, 1: 2000), FANCL (A6812, ABclonal, 1: 2000), FANCB (A2435, ABclonal, 1: 2000), FAAP100 (A8591, ABclonal, 1: 2000), FANCA (A9529, ABclonal, 1: 2000), FANCC (A1812, ABclonal, 1: 2000), FANCF (18060-1-AP, Proteintech, 1: 2000), FANCG (A6206, ABclonal, 1: 2000), FANCM (A7602, ABclonal, 1: 2000), FAAP24 (26431-1-AP, Proteintech, 1: 2000), p-SMAD2 (80427-2-RR, Proteintech, 1: 2000), SMAD2 (67343-1-Ig, Proteintech, 1: 2000), p-STAT1 (82674-10-RR, Proteintech, 1: 2000), STAT1 (66545-1-Ig, Proteintech, 1: 2000), p-ERK1/2 (80031-1-RR, Proteintech, 1: 2000), ERK1/2 (11257-1-AP, Proteintech, 1: 2000). After washing, membranes were incubated with HRP-conjugated goat anti rabbit IgG (SA00001-2, Proteintech, 1: 5000) or HRP-conjugated goat anti-mouse IgG (SA00001-1, Proteintech, 1: 5000) for 1 h at room temperature. Protein bands were visualized using BeyoECL Plus (P0018S, Beyotime) and imaged with a ChemiDoc XRS + system (Bio-Rad, USA).
Immunofluorescence Assay
1 × 105 cells were seeded onto glass coverslips in 24-well plates and cultured until approximately 70% confluency. After treatment, cells were fixed with 4% paraformaldehyde for 15 minutes at room temperature and permeabilized with 0.1% Triton X-100 in PBS for 10 min. The samples were then blocked with 5% bovine serum albumin (BSA) in PBS for 1 h and incubated with primary antibodies STAT1 (1: 500) or p-STAT1 (1: 200) overnight at 4°C. After washing with PBS, cells were incubated with CoraLite488-conjugated goat anti-mouse IgG (SA00013-1, Proteintech, 1: 10 000) or CoraLite594-conjugated donkey anti-rabbit IgG (SA00013-8, Proteintech, 1: 10 000) for 1 h at room temperature in the dark. Coverslips were mounted with antifade mounting medium containing with DAPI (P0131, Beyotime) and visualized under a Zeiss Axio Observer 7 (Zeiss, Germany). The number of nuclear foci was quantified using ImageJ.
Immunoprecipitation (IP)
IP assays were performed as previously described.24-26 5 × 106 cells were lysed in IP lysis buffer (P0013, Beyotime) containing protease and phosphatase inhibitors on ice for 30 minutes. The lysates were centrifuged at 12 000 × g for 15 min at 4°C to remove debris. For each immunoprecipitation, 1 mg of total protein was incubated with 1 μg of primary antibody against FANCD2 overnight at 4°C with gentle rotation. The next day, 20-30 μL of Pierce protein A/G agarose beads (20 422, Thermo Fisher Scientific, USA) were added and incubated for an additional 2 h at 4°C. Beads were washed 5 times with lysis buffer, and the bound proteins were eluted by boiling in SDS sample buffer. Samples were subjected to SDS-PAGE and analyzed by western blotting using the indicated antibodies.
Chromatin Immunoprecipitation (ChIP)
ChIP assays were performed using the SimpleChIP Enzymatic Chromatin IP Kit (9003, Cell Signaling Technology, USA) according to the manufacturer’s instructions. Briefly, 5 × 106 cells were crosslinked with 1% formaldehyde for 10 min at room temperature, followed by quenching with 125 mM glycine. Nuclei were isolated and chromatin was digested with micrococcal nuclease and further sonicated to obtain DNA fragments of approximately 200-500 bp. The chromatin solution was immunoprecipitated overnight at 4°C with 1 μg STAT1 antibody (9172, Cell Signaling Technology) or 1 μg rabbit IgG (2729, Cell Signaling Technology). Pierce protein A/G agarose beads were used to capture immune complexes, followed by sequential washing, elution, and reverse crosslinking at 65°C for 2 h. DNA was purified using spin columns provided in the kit and analyzed by quantitative PCR using gene-specific primers targeting promoter regions of interest. ChIP-qPCR enrichment was calculated as % input relative to IgG control.
Quantitative Polymerase Chain Reaction (qPCR)
Total RNA was extracted using Super FastPure Cell RNA Isolation Kit (RC102-01, Vazyme, China). RNA quality was verified by A260/280 = 1.8-2.1 and intact rRNA peaks; 1 µg RNA was reverse-transcribed using random hexamers in a 20 µL reaction by HiScript IV 1st Strand cDNA synthesis Kit (R412-01, Vazyme). cDNA template was diluted 1:10 before preparing PCR system. qPCR used a 20 µL reaction (10 µL 2× SupRealQ Purple Universal SYBR qPCR Master Mix (Q713-02, Vazyme), 0.4 µL each primer at 10 µM, 2 µL cDNA, nuclease-free water to volume) on a 96-well instrument with: 95°C 2 min; 40 cycles of 95°C 15 s, 60°C 60 s; followed by melt-curve (65-95°C). Primers were designed to yield 200 bp amplicons and to span exon-exon junctions where possible to minimize genomic DNA amplification. Each sample was run in technical triplicate; no-RT and no-template controls were included. Expression was normalized to GAPDH and analyzed by the 2^-ΔΔCt method, reporting MIQE-compliant details. Primer sequences were listed below:
FANCA, F: TGGAGAGCTGGTCCTTCCATTC, R: CTTTCTCTTGCTTCGGCTTCTC;
FANCB, F: TTCCTGCCAGTGTCTCTGACAT, R: CTGGCTCATCCTTCAGGTAGGT;
FANCC, F: CTGGCTCCAGACACTGAAGCAT, R: ATTGCTCTGCCACCATCTCAGC;
FANCF, F: TGCACAACCAGTGGAGGCAAGA, R: GAGTTGCTGCACCAGGTGGTA;
FANCG, F: GAGAGTCTGGAGCTGCTAGTTG, R: TGTGCTTGGTCTGGCTCTGAGT;
FANCL, F: GGAGTGCAACAGCACGCAGAAT, R: CTGCTCAGCTTAATTCCCAGGG;
FANCM, F: TCCAGCATAGTTTGCCGTCACT, R: GCTTGGTCTCTCAGGGTTCTGG;
FAAP100, F: TGCTGTCTGGAATTGGCAAC, R: GCTTGTCAGTGCCTTGTTCC;
FAAP24, F: ACCGAAGCTGATTTGGTGGCAG, R: CCATTCCAAGGTCCAGCACAGT.
FANCA for ChIP, F: AAGTGATGGGGTCTCGCTCTGTT, R: TCTGAGCTCCGGGCAGCCGCTGT;
FANCC for ChIP, F: TTACAGGCTGAGCCACTGCGCATGT, R: TGCCCCGGCTCCTGCCCCGGCTC;
FANCF for ChIP, F: CTACTTAAGGATATTTCCAAAG, R: TTTCACCTTGGAGACGGCGACTCTCTG;
FANCM for ChIP, F: CATTGTCCCCGGGTATGTAATATTT, R: CCAGTTTATCCAGTTGAAAATGCCGC.
Xenograft Tumor Model
Male BALB/c nude mice (4-6 weeks old, 18-22 g) were purchased from (Aniphe BioLab, China) and housed under specific pathogen-free (SPF) conditions with free access to food and water. All animal experiments were approved by the Institutional Animal Care and Use Committee of Nanjing Medical University (HJSW-23100801). Three groups (n = 5) were defined as “NC”, “cisplatin” and “cisplatin plus TSN”. 5 × 108 T24 bladder cancer cells suspended in 100 μL of PBS were subcutaneously injected into the right flank of each mouse. Tumor volume was measured every 3 days using a digital caliper a nd calculated using the formula: Volume = (length × width2)/2. When tumors reached approximately 100 mm3, mice were randomly divided into treatment groups and administered with toosendanin by intraperitoneal injection at 5 mg/kg daily as well as cisplatin by intraperitoneal injection at 3 mg/kg every 3 days. Mice were monitored for body weight and tumor growth until euthanasia. Tumors were excised, weighed, and subjected to further analysis, including western blotting.
Cell Counting Kit-8 (CCK-8) Assay
Cells were added 10 µL/well CCK-8 (HY-K0301, MedChemExpress), then incubated at 37°C for 1 h. Cell viability was measured under the absorbance at 450 nm, and values were blank-subtracted and normalized to the Vehicle/No-UV group (set to 100%). Experimental groups were: Vehicle/No-UV, TSN/No-UV, Vehicle/+UV, and TSN/+UV. Each condition included 3 technical replicates and experiments were repeated at least 3 times independently.
Statistical Analysis
All experiments were performed at least 3 times independently. Data were presented as mean ± standard deviation (SD). Statistical analyses were conducted using GraphPad Prism 8.0 software (GraphPad Software, USA). Comparisons between 2 groups were performed using an unpaired two-tailed Student’s t-test. For multiple group comparisons, one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was applied. Two-way ANOVA was used to analyze the significant interaction between TSN and UV effects. P-value less than 0.05 was considered as statistical significance.
Results
Inhibition of FANCI/D2 Activation by Toosendanin in Bladder Cancer Cells
Three bladder cancer T24, RT4 and J82 cell lines were exposed to ultraviolet radiation c (UVC) to trigger the activation of Fanconi anemia (FA) signaling pathway. Although these cell lines represent different origins, p53 genotypes and different differentiation grades as known to all, monoubiquitination of both FANCI and FANCD2 (top band) was observed upon UVC stimulation (Figure 1A-C), indicating that the activation mechanism of the FA signaling pathway was conserved in the bladder cancer cell line. However, pretreatment with toosendanin (TSN) followed by UVC exposure abolished the monoubiquitination of FANCI and FANCD2 (Figure 1A-C). FANCD2 foci by immunofluorescence were weakened by TSN pretreatment (Figure 1D-I). The interaction between ID2 and SLX4 (FANCP) by immunoprecipitation was also diminished in bladder cancer cells (Figure 1J-L), implying that the recruitment process of the following DNA repair factors (such as SLX4/XPF/ERCC1 for removing the damaged chain of interstrand crosslinks) was disrupted by TSN. Now, we determined that TSN was capable of inhibiting FA pathway activation in bladder cancer in vitro. Toosendanin inhibits the activation of FA pathway. (A-C) Western blot shows the monoubiquitination of FANCI and FANCD2 in bladder cancer T24 (A), RT4 (B) and J82 (C) cell lines induced by UVC and treated with TSN. Top band is the monoubiquitinated FANCI or FANCD2, and the bottom band is the ones that have not undergone monoubiquitination. (D-I) Immunofluorescence shows the FANCD2 staining indicating the foci in bladder cancer T24 (D), RT4 (E) and J82 (F) cell lines induced by UVC and treated with TSN. Scale bar, 10 μm. One-way ANOVA shows the quantitative differences of FANCD2 foci among different groups of T24 (G), RT4 (H) and J82 (I) cell lines. Numerical P-values indicate the statistical significance compared with NC and UV groups respectively. (J-L) co-IP shows the interaction among FANCI, FANCD2 and SLX4 in T24 (J), RT4 (K) and J82 (L) cell lines. Abbreviation: NC, negative control; UV, UVC; T, TSN, mub, mono-ubiquitination
Down-Regulation of FA Core Subunits by Toosendanin via Suppressing JAK/STAT1 Pathway
Next, we further examined the expression of FA core complex components representing distinct functional modules: the catalytic E3 ligase module (FANCL, FANCB, FAAP100), the structural/assembly module (FANCA, FANCC, FANCF, FANCG), and the DNA recognition module (FANCM, FAAP24). We observed that the expression of most assembly module components, except FANCG, was significantly reduced, and FANCM expression also showed a decrease. In contrast, the expression of FANCL, FANCB, FAAP100 and FAAP24 remained robust and unaffected by TSN treatment (Figure 2A-C). RNA levels of FANCA, FANCC, FANCF and FANCM also showed a consistent decline by TSN (Figure 2D-F), indicating that TSN impaired FA pathway activation through down-regulating a part of FA components at the transcriptional level. Toosendanin down-regulates FA subunits. (A-C) Western blot shows the expression of FANCL, FANCB, FAAP100, FANCA, FANCC, FANCF, FANCG, FANCM and FAAP24 in bladder cancer T24 (A), RT4 (B) and J82 (C) cell lines induced by UVC and treated with TSN. (D-F) Histogram shows the RNA levels of FANCL, FANCB, FAAP100, FANCA, FANCC, FANCF, FANCG, FANCM and FAAP24 in bladder cancer T24 (D), RT4 (E) and J82 (F) cell lines induced by UVC and treated with TSN by qPCR. Numerical P-values indicate the statistical significance compared with cells treated with UVC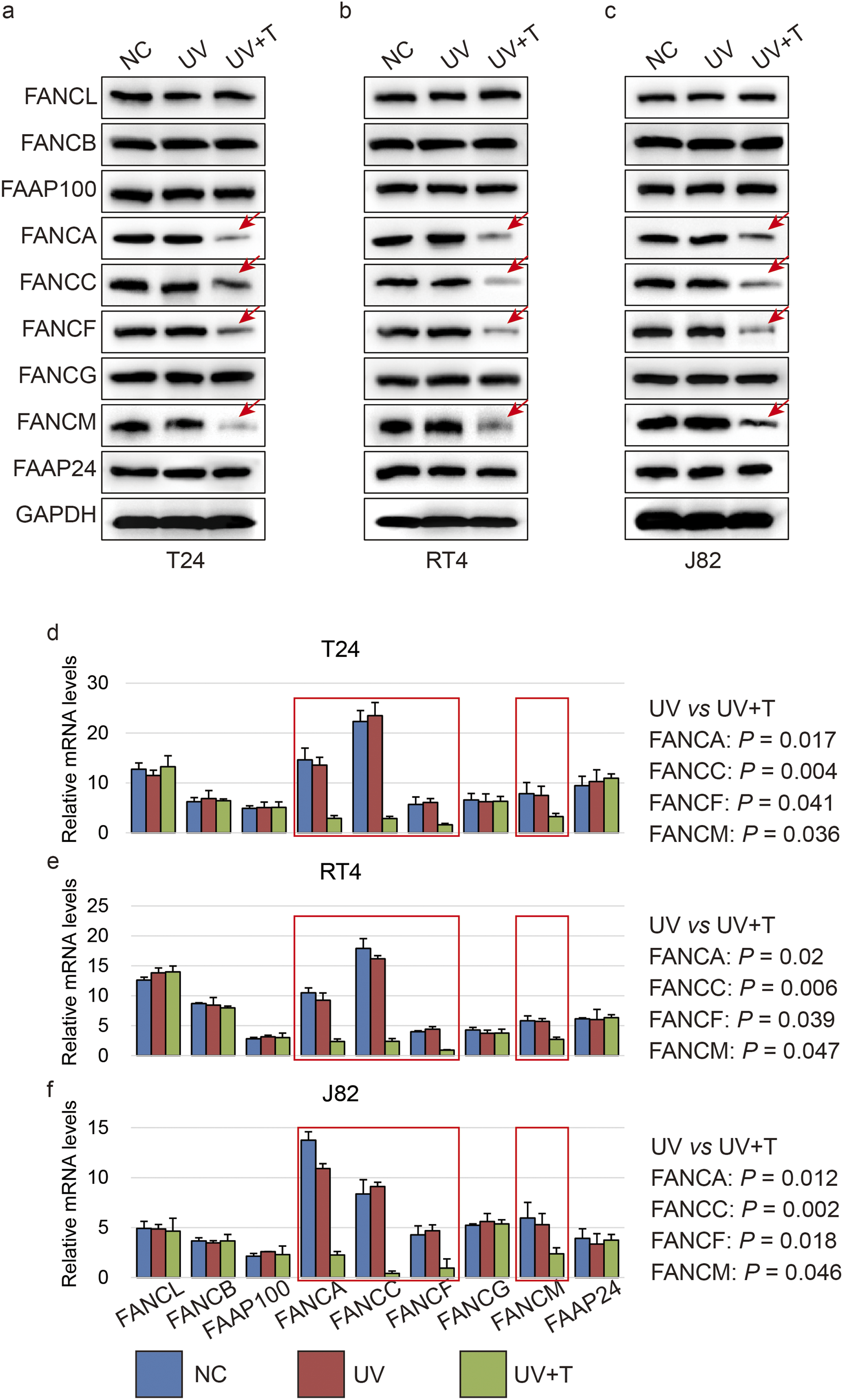
As previously described, activation of TGF-β/SMAD2,
27
JAK/STAT1
28
and MAPK/ERK (possible)
29
signaling pathways were implicated in enhancing expression of FA subunits. Herein, TGF-β/SMAD2 was indeed activated by UVC, but unaffected by TSN. MAPK/ERK was seemingly not involved with UVC stimulation in our system. Phosphorylation of STAT1 was elevated by UVC, and substantially reduced by TSN (Figure 3A-C). The nuclear translocation of p-SMAD2, p-STAT1 and p-ERK was also validated in T24 cells by immunofluorescence, and only JAK/STAT1 was indeed inactivated by TSN (Figure 3D-F). Furthermore, the agonist and antagonist of JAK/STAT1 were employed respectively. JAK/STAT1 antagonist could mimic the effect of TSN on the expression of FANCA, FANCC, FANCF and FANCM, and JAK/STAT1 agonist could compromise the TSN’ effect on the down-regulation of these FA genes (Figure 3G-I). Consistently, transcripts of FANCA, FANCC, FANCF and FANCM were substantially rescued by JAK agonist (Figure 3J-L), and ChIP-qPCR assay also determined that STAT1 binding capacity on promoters of these 4 genes was weakened by TSN compared to UVC, but rescued by JAK agonist (Figure 3M-O). Collectively, we determined that TSN suppressed FA pathway activity via inhibiting JAK/STAT1 pathway. Toosendanin inhibits JAK/STAT1 pathway. (A-C) Western blot shows the phosphorylation of SMAD2, STAT1 and ERK in bladder cancer T24 (A), RT4 (B) and J82 (C) cell lines induced by UVC and treated with TSN. (D-F) Immunofluorescence shows the nuclear translocation of p-SMAD2 (D), p-STAT1 (E) and p-ERK (F) in bladder cancer T24 cells induced by UVC and treated with TSN. (G-I) Western blot shows the phosphorylation of STAT1 and the expression of FANCA, FANCC, FANCF and FANCM in bladder cancer T24 (G), RT4 (H) and J82 (I) cell lines induced by UVC and treated with TSN, JAK/STAT1 agonist or inhibitor. (J-L) Histogram shows the RNA levels of FANCA, FANCC, FANCF and FANCM in bladder cancer T24 (J), RT4 (K) and J82 (L) cell lines induced by UVC and treated with TSN as well as JAK/STAT1 agonist and inhibitor by qPCR. (M-O) ChIP-qPCR shows the binding capacity of STAT1 on promoters of FANCA, FANCC, FANCF and FANCM in bladder cancer T24 (M), RT4 (N) and J82 (O) cell lines induced by UVC and treated with TSN and JAK/STAT1 agonist. Numerical P-values indicate the statistical significance compared with cells treated with UVC and TSN. Abbreviation: JI, JAK/STAT1 inhibitor Ruxolitinib; JA, JAK/STAT1 agonist IFN-γ
Cytotoxic Effect of Platinum-Based Drugs on Bladder Cancer Facilitated by Toosendanin
Given that the FA signaling pathway is crucial for repairing DNA damage caused by platinum-based and other anticancer agents, TSN holds significant potential in amplifying the cytotoxic effects of radiotherapy and chemotherapy. A xenograft mouse model generated by subcutaneous injection of T24 cells was intraperitoneally administrated with TSN and cisplatin. We observed that tumor size was significantly reduced in the groups treated with TSN compared to those without TSN (Figure 4A). The monoubiquitination of FANCI and FANCD2 was compromised by TSN (Figure 4B). The expressions of FANCA, FANCC, FANCF and FANCM were consistently reduced by TSN (Figure 4B). The phosphorylation of STAT1 was also weakened by TSN (Figure 4B). Toosendanin enhances the cisplatin-induced cytotoxic effect on bladder cells in vivo. (A) Comparison of tumor sizes among groups of regular T24 cells, T24 cells treated with cisplatin, as well as T24 cells treated with cisplatin and TSN (top). One-way ANOVA shows the significant differences compared to NC and UV indicated by numerical P-values (bottom). (B) Western blot shows the expression of FANCI, FANCD2, FANCA, FANCC, FANCF and FANCM as well as the phosphorylation of STAT1 in xenograft tumor of regular T24 cells, T24 cells treated with cisplatin, as well as T24 cells treated with cisplatin and TSN. Abbreviation: Cis, cisplatin; T, TSN; mub, mono-ubiquitination; Tyr701, 701st tyrosine phosphorylation
Selective Enhancement of Cisplatin Sensitivity by Toosendanin in Bladder Cancer Cell Lines
To validate the probable selectivity of TSN-mediated sensitization in bladder cancer, we extended our experiments to include a non-malignant urothelial cell line (SV-HUC-1) and a non-bladder cancer line (A549) alongside T24 cells. In single-agent assays, although TSN showed relatively lower cytotoxicity in SV-HUC-1 cells than in T24 and A549 cells, its high IC50 values across all cell lines indicate minimal cytotoxic effects under the tested conditions (Figure 5A). UV exposure alone reduced viability to 59% in bladder cancer cells and 82% in SV-HUC-1 and 76% in A549 cells. Notably, TSN (100 nM) pre-treatment could further decrease viability to 27% in T24 cells, while have little additional effect in SV-HUC-1 (76%) and A549 (64%) (Figure 5B). Importantly, two-way ANOVA confirmed a significant interaction between TSN and UV treatment in T24 cells (P = 0.007), indicating a synergistic sensitization effect, while no significant interaction was observed in SV-HUC-1 or A549 (Figure 5C). These data strongly supported the tumor-selective nature of TSN in sensitizing bladder cancer cells to UV exposure. Toosendanin selectively enhances UV-induced cytotoxicity in bladder cancer cells. (A) Dose-response curves of TSN (10, 20, 40, 80, 160, 320 nM for 24 h) as a single agent in T24 cells, the non-malignant urothelial cell line (SV-HUC-1), and a non-bladder cancer line (A549). TSN exhibited markedly lower cytotoxicity in SV-HUC-1 cells compared to T24 and A549 cells. (B) Cell viability (mean ± SEM, n = 3 biological replicates) in T24, SV-HUC-1, and A549 cells under 4 conditions: Untreatment, TSN, UV, and UV + TSN. One-way ANOVA shows the significant differences compared to UV + TSN indicated by numerical P-values. (C) Two-way ANOVA interaction plots demonstrating significant TSN × UV effects in bladder cancer cells (F = 12.88, P = 0.007) but no interaction in SV-HUC-1 or A549 (P > 0.05)
Taken together, we determined that TSN played an inhibitory role in FA signaling pathway via suppressing JAK/STAT1 in bladder cancer.
Discussion
The FA signaling pathway is a critical DNA repair mechanism dedicated to the resolution of DNA interstrand crosslinks, a particularly cytotoxic type of DNA lesion that can be induced by chemotherapeutic agents such as cisplatin and mitomycin C.30,31 Central to this pathway is the FANCI-FANCD2 heterodimer, which is first recruited to stalled replication forks or interstrand crosslink sites and binds the damaged DNA. The FA core complex, functioning as an E3 ubiquitin ligase, subsequently catalyzes the monoubiquitination of both FANCI and FANCD2. This post-translational modification stabilizes the ID2 complex on chromatin and promotes the recruitment of downstream nucleases, translesion synthesis polymerases, and homologous recombination factors required for complete ICL repair. 32 Increasing evidence indicates that hyperactivation of the FA pathway contributes to chemoresistance in various malignancies,33,34 including bladder cancer. 35 This underscores the need for therapeutic strategies that can effectively inhibit FA pathway activation to enhance the efficacy of DNA-damaging agents.
In our study, we used UVC irradiation to induce DNA damage in bladder cancer cell lines and monitored FA pathway activation by assessing FANCI and FANCD2 monoubiquitination. UVC exposure successfully triggered ID2 activation in T24, RT4, and J82 cells, as evidenced by increased levels of monoubiquitinated FANCI and FANCD2. However, pretreatment with TSN significantly diminished this monoubiquitination. These findings suggest that TSN interferes with a critical step in FA signaling, potentially impairing the repair of DNA damage and sensitizing cells to genotoxic stress. Beyond biochemical analysis, immunofluorescence staining revealed that UVC-induced FANCD2 nuclear foci - indicative of active DNA repair sites - were markedly reduced upon TSN treatment. This reduction was consistent across all tested bladder cancer cell lines. In addition, co-immunoprecipitation assays demonstrated that TSN impaired the physical interaction between ID2 and SLX4. Since the ID2 complex is essential for downstream DNA repair processes, its disruption by TSN implies that TSN not only blocks monoubiquitination but also compromises FA signaling fidelity and function. To elucidate the molecular basis of TSN-mediated FA pathway inhibition, we examined the expression of FA core complex subunits. Interestingly, TSN significantly down-regulated the mRNA and protein levels of several key components, particularly the FA core complex subunits (FANCA, FANCC, FANCF, FANCG, and FANCM). These components are crucial for the structural integrity and catalytic function of the FA core complex. The expression of FANCL, FANCB, and FAAP100, while essential, remained relatively unaffected, suggesting that TSN specifically targets transcriptional regulation of structural elements of the complex rather than all components indiscriminately.
We next sought to uncover the upstream signaling mechanisms responsible for FA core gene down-regulation by TSN. Literatures and our observation both pointed to the JAK/STAT1 axis as a key regulator for FA genes’ transcription. STAT1 is known to transcriptionally regulate DNA repair genes under stress conditions. 36 We found that TSN treatment significantly inhibited the phosphorylation of STAT1, an essential step for its nuclear translocation and transcriptional activity. Concomitantly, expression levels of FA core genes were suppressed, confirming that JAK/STAT1 inhibition mediates TSN’s effect on FA pathway attenuation. To verify our in vitro findings in an in vivo context, we established a xenograft tumor model using T24 bladder cancer cells. Mice treated with TSN exhibited significantly reduced tumor growth compared to controls, supporting the anti-tumor efficacy of TSN. Western blot analysis of tumor tissues confirmed that TSN suppressed the expression of FA core complex genes and reduced monoubiquitination of FANCI and FANCD2 in vivo. These results validate the biological relevance of our cellular findings and indicate that TSN can effectively inhibit FA pathway activation in a tumor microenvironment. This finding is particularly intriguing as it links an immune-modulatory pathway with DNA repair machinery, providing a dual-targeting rationale. It also opens avenues for combining TSN with JAK inhibitors or DNA-damaging agents in therapeutic regimens.
Given that many chemotherapeutic agents used to treat bladder cancer, such as cisplatin, function through inducing DNA damage, the FA pathway serves as a critical determinant of treatment resistance. Our results demonstrate that TSN effectively impairs FA pathway activation by blocking JAK/STAT1-mediated transcription of core complex components. This renders tumor cells more susceptible to DNA damage and may enhance the efficacy of existing chemotherapeutic regimens. Moreover, TSN’s ability to specifically down-regulate scaffold subunits of the FA core complex is noteworthy, as it allows for selective disruption of the pathway without complete suppression of DNA repair capacity, potentially minimizing systemic toxicity. Despite the promising findings presented in this study, several limitations should be acknowledged. First, while we demonstrated that TSN treatment reduced the monoubiquitination levels of FANCI and FANCD2, and diminished their nuclear foci formation and interaction, the functional consequences of these changes on DNA repair capacity were not directly assessed. Without functional assays such as cell survival under mitomycin C or cisplatin treatment, or chromosome breakage analysis, it is difficult to conclude that TSN functionally impairs the FA DNA repair pathway. Future studies should incorporate these assays to confirm the biological significance of FA pathway inhibition. Second, although we identified the JAK/STAT1 signaling pathway as a potential upstream regulator of several core FA components (including FANCA, FANCC, FANCF, FANCG, and FANCM), the direct transcriptional regulatory relationship remains to be fully validated. Our conclusions are primarily based on the observed changes in gene and protein expression, which may be indirect. Future work should perform luciferase reporter assays and ChIP experiments to determine whether STAT1 directly binds to and regulates the promoters of these FA genes. Third, the in vivo evidence provided in this study was limited to tumor growth inhibition and reduced monoubiquitination of FA proteins in xenograft tumors. The enhancement of cisplatin efficacy or DNA damage accumulation in vivo remains to be examined. 37 Expanding the in vivo model to include combination treatments and assessing additional markers of DNA damage, such as FANCD2 foci, would help clarify the therapeutic potential of TSN as a chemo-sensitizer. Fourth, the selectivity and safety of TSN were not fully evaluated. While we observed potent effects on bladder cancer cells, the impact on normal urothelial cells was not tested. Assessing cytotoxicity and FA pathway suppression in non-tumorigenic bladder epithelial cell lines would provide a better understanding of TSN’s therapeutic window and specificity. Moreover, our experimental models are still largely restricted to bladder cancer cell lines. Although this choice was guided by the high clinical relevance of the FA pathway to cisplatin-based chemotherapy in bladder cancer, these findings should not be interpreted as indicating bladder cancer-specificity. We only expand our observation to SV-HUC-1 and A549 cells. TSN may exert similar effects on the FA pathway in other tumor types, as the pathway is broadly conserved across cancers; however, this possibility remains to be investigated in future studies. Finally, our findings raise intriguing questions regarding the broader role of TSN in regulating other DNA repair pathways. It is plausible that TSN may interfere with additional repair mechanisms beyond the FA pathway, such as homologous recombination or nucleotide excision repair. Multi-omics approaches, including RNA-seq and ATAC-seq, could be applied to map the full extent of TSN-induced changes in DNA damage response networks.
Future work should investigate whether TSN acts synergistically with platinum-based drugs in resistant bladder cancer models and explore the selectivity of TSN’s action in tumor vs normal urothelial cells. Elucidating the direct targets of TSN within the JAK/STAT1 pathway will also be crucial for its further development as a targeted therapy.
Conclusion
TSN inhibits FA DNA repair signaling in bladder cancer by suppressing JAK/STAT1-mediated FA core gene transcription, supporting its potential as a combinatorial agent to overcome cisplatin resistance.
Footnotes
Author Contributions
Z.W. performed all cellular and molecular experiments, and analyzed the raw data. X.Z. helped assist animal study. Z.L. helped revise manuscript. L.W. designed the overall project, provided the financial support, supervised the entire experimental process and drafted the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research is financially supported by Hospital-level project of Taizhou People’s Hospital (ZL201938).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Data will be made available on request.