
Abstract
Objectives
The intestine exhibits high radiosensitivity and is susceptible to damage during radiotherapy for abdominal or pelvic tumors, especially at radiation doses exceeding 5 Gy, which can lead to severe gastrointestinal complications. This study aims to elucidate the mechanisms of DNA repair and immune responses in radiation-induced intestinal injury, with the goal of enhancing radiotherapy efficacy.
Methods
We employed weighted correlation network analysis (WGCNA) to analyze RNA sequencing data from intestinal tissues collected at various time points following irradiation, as well as from mice with targeted disruption of the NFE2-like bZIP transcription factor 2 (Nrf2) gene and their wild-type counterparts.
Results
Our analysis revealed enhanced DNA repair capacity and attenuated immune response 3.5 days after irradiation. Histone variants and ribosomal proteins were identified as potential contributors to DNA repair processes. We also constructed a competing endogenous RNAs (ceRNA) network including genes from both the DNA repair and immune modules and identified Akr1b8, Gsta3, and Nqo1 as radiation-effect genes regulated by Nrf2.
Conclusions
These findings provide valuable insights into the molecular mechanisms of radiation-induced intestinal injury and suggest potential targets for protecting normal intestinal tissue during radiotherapy. This knowledge may contribute to improved treatment outcomes and enhanced patient well-being.

Introduction
At least 50% of cancer patients undergo radiotherapy, which contributes to 25% of cancer cures. 1 Despite recent advancements in treatment technology, radiation toxicity in normal tissues remains a significant barrier to effective cancer treatment in patients with localized disease. 1 During radiotherapy for abdominal or pelvic tumors, the highly radiosensitive intestine is particularly vulnerable. 1 Gastrointestinal damage occurs when the entire intestine receives a radiation dose exceeding 5 Gy. 2 Approximately 50% of patients receiving abdominal or pelvic radiotherapy experience chronic intestinal dysfunction to varying degrees, with side effects such as vomiting, weight loss, anorexia, dehydration, diarrhea, and infection. 2 In severe cases, these effects can lead to intestinal necrosis, intestinal perforation, and even death. 2 Symptoms of radiation enteropathy may emerge within hours (early), weeks (acute), or even months to years (delayed or chronic) following radiotherapy. 3
Ionizing radiation (IR) induces DNA damage through reactive oxygen species (ROS) generated by water ionization within cells. 4 The transcription factor NFE2-like bZIP factor 2 (Nrf2), renowned for its antioxidant properties, plays a pivotal role in scavenging excessive ROS that accumulate after irradiation. 5 IR can also directly cause DNA damage, including base modifications, single-strand breaks (SSBs), and double-strand breaks (DSBs). 4 Recent studies have revealed the involvement of Nrf2 in the base excision repair (BER), homologous recombination (HR), and non-homologous end joining (NHEJ) pathways, highlighting its capacity to facilitate the repair of DNA damage induced by radiation.6-8 The DNA damage response initially induces reversible cell cycle arrest, providing cells with time to repair. 4 When DNA damage cannot be adequately repaired, the DNA damage response may shift towards apoptosis or senescence signaling pathway. 4 The intestine, as the largest immune organ in the body, plays a vital role in maintaining immune homeostasis. 2 The intestinal immune system, comprising gut-associated lymphoid tissue and secretory immunoglobulins, recognizes intestinal flora and antigens and responds in distinct ways to maintain both intestinal and systemic immune balance. 2
Understanding the mechanisms of DNA repair and immune responses in radiation-induced intestinal injury is crucial for improving radiotherapy efficacy and patients’ quality of life. In light of the emerging role of Nrf2 in DNA repair, it is of particular interest to explore the underlying mechanisms by analyzing the transcriptome of Nrf2 knockout mice. To this end, we performed weighted correlation network analysis (WGCNA) on the sequencing data from intestinal tissues at various time points post-irradiation, as well as from Nrf2 knockout and wild-type mice. This analysis aims to explore the mechanisms of alternative splicing and the competing endogenous RNAs(ceRNA) network in radiation effects, identify key genes at different time points after irradiation, and screen for radiation effect genes potentially regulated by Nrf2.
Materials and Methods
Sample Preparation and RNA Sequencing
Six to eight-week-old male C57BL/6 mice were purchased from Beijing Huafukang Experimental Animal Technology Co., Ltd. The animal research protocol was reviewed and approved by the Ethics Committee of the Institute of Radiation Medicine, Chinese Academy of Medical Sciences (Approval No. IRM-DWLL-20160201; Approval date: 2016-05-15). In March 2017, mice were divided into four groups, with three mice in each of the 0 Gy group (control group), 6 h post-irradiation group, 3.5 days after irradiation group, and 5 days after irradiation group for the first transcriptome sequencing. In August 2017, mice were divided into four groups, with three mice in each of the WT group, WT_IR group, Nrf2_KO group, and Nrf2_KO_IR group for the second transcriptome sequencing. The mice were irradiated with full abdomen at room temperature, with an irradiation dose of 14 Gy. The irradiation source was a 137Cs γ-ray irradiation source provided by the Institute of Radiation Medicine, Chinese Academy of Medical Sciences. Mice were sacrificed at different time points after irradiation, and jejunal tissue was collected to extract total RNA using the TRIZOL method. RNA library preparation and sequencing were completed by Cloud-seq Biotech Inc.
Weighted Gene Co-Expression Network Analysis (WGCNA)
The co-expression network was constructed using the WGCNA (1.72-5) package in R. 9 In the analysis of the first sequencing data, power was 5, TOMType was unsigned, mergeCutHeight was 0.25, and minModuleSize was 30. Genes were clustered into 51 related modules. In the analysis of the second sequencing data, power was 9, TOMType was unsigned, mergeCutHeight was 0.25, and minModuleSize was 30. Genes were clustered into 32 related modules. Subsequently, GO and KEGG pathway enrichment analysis was performed on the adjacent genes of lncRNA and circRNA and the genes of coding RNA in the module to analyze the biological functions.
Construction of ceRNA and Protein-Protein Interaction (PPI) Network
The predicted target miRNAs of circRNA were provided by Cloud-seq Biotech Inc; the sequences of lncRNA were obtained from UCSC (genome.ucsc.edu), and the target miRNAs were predicted using the miRDB database (mirdb.org), with those scoring greater than 80 selected10,11; the target miRNAs of coding RNA were from the ENCOLI (rnasysu.com/encori) database. 12
The DESeq2 (1.38.3) package was used to screen for differentially expressed genes, and immune-related genes with log2foldchange greater than 0.5 and repair-related genes with log2foldchange greater than 0.8 were selected. The PPI network was constructed using STRING (cn.string-db.org) and plotted using cytoscape. 13
Alternative Splicing Analysis
The mouse transcriptome data were obtained from GENECODE (gencodegenes.org/mouse), and the index was constructed using the Rsubread package. 14 The edgeR (3.40.2) package was used to analyze the bam file and screen the differentially expressed exons with |log2foldchange|>0.8 in different groups. 15 The upset graph was drawn using the UpSetR (1.4.0) package. 16
Immune Infiltration Analysis
The immune infiltration levels of the samples were calculated using the CIBERSORT (0.1.0) package and the mMCPcounter (1.1.0) package, respectively.17,18 The mouse immune cell expression profile was referenced from Chen et al 19 Kruskal test was used to determine whether the difference in invasion between groups was significant.The WGCNA package was used to analyze the correlation between the immune cell infiltration level and the module. The GSVA (1.46.0) package was used to score the merged immune-related genes. 20
From March to May 2018, male C57BL/6 mice were divided into three groups: 0 Gy group (Ctrl group), 3.5 days post-irradiation group, and 5 days post-irradiation group, with three mice in each group. Mice were irradiated with a whole abdomen of 14 Gy at room temperature and sacrificed at different time points after irradiation. The small intestinal lamina propria suspension was prepared using the lamina propria isolation kit (130-097-410, Miltenyi Biotec, Germany) according to the manufacturer’s protocol. The following antibodies were used to stain the antigens on the single cell suspension: CD3 (100306, FITC, Biolegend, USA), CD4 (100516, APC, Biolegend), and CD8 (100708, PE, Biolegend, USA). The stained cells were then analyzed by flow cytometry in the FITC, APC, and PE channels (Mindray, China). CD3+CD4+ cells are CD4 T cells, and CD3+CD8+ cells are CD8 T cells. Antigen staining was performed on single cell suspensions using the following antibodies: CD45 (103107, FITC, Biolegend, USA), I-Ab (116415, PerCP, Biolegend), CD103 (121413, APC, Biolegend, USA), and CD11c (117307, PE, Biolegend, USA). Stained cells were then analyzed by flow cytometry in the FITC, PerCP, APC, and PE channels (Mindray, China) to distinguish DCs from macrophages according to the protocol of Geem et al 21 Data analysis was performed using Flowjo software (TreeStar, Ashland, OR, USA). Graphpad was used for significance analysis and graphing. An unpaired t-test was used to determine whether there was a significant difference in invasion between the groups.
Single-Cell Sequencing Data Analysis
Single-cell RNA-seq (scRNA-seq) data were processed using Seurat (v.5.1.0). 22 Cells expressing at least 500 genes but fewer than 7000 genes, and with mitochondrial gene content <40%, were retained for downstream analysis, resulting in a total of 4407 cells.
In Seurat, normalized data were first scaled, and samples from the entire epithelium, both normal and irradiated, were combined into a single data set. Principal components were calculated based on all genes in the dataset, and the main principal components were identified. Cells were then grouped into unsupervised clusters using a shared-nearest-neighbour modularity optimization-based clustering algorithm (Smart Local Moving algorithm). The optimal clustering resolution was determined using the clustree package by plotting clustering trees with resolutions ranging from 0 to 4 in 0.5 increments.
Cells were visualized using the t-SNE method. Differentially expressed genes for each cluster were identified using the Wilcox test in Seurat, and cell types were annotated based on the top genes (P value <0.05, |logFC| >0.25). Cell type annotations referred to SingleR package annotations and characteristic genes for cell subpopulations reported by Ayyaz et al 23
Cells were classified into seven groups: Enterocyte cell (EC), Crypt-base columnar cell (CBC), Goblet cell (GC), Lymphocyte cell (LC), Enteroendocrine cell (EE), Paneth cell (PC), and Tuft cell (TC). DNA repair gene scores for each cell group were analyzed using the msigdbr and GSVA packages. The Wilcox test was used to analyze the significance of differences.
Results
Radiation Effects on Intestinal Tissue at Different Time Points after Irradiation
Our team sequenced mouse intestinal tissues at various time points post-irradiation to analyze the potential ceRNA network.24-26 WGCNA is a powerful tool for analyzing RNA sequencing data, offering deep insights into gene co-expression networks, functional modules, and their relationships with phenotypic traits. Therefore, we reanalyzed these sequencing data using WGCNA and identified modules (gene sets) significantly associated with each time point (Figure 1). Among them, the blue module showed a strong positive correlation with the 6-h post-irradiation group. GO analysis indicated that this module is related to the regulation of proliferation (GO: 0030336), cell movement (GO: 0040013), and autophagy (GO: 0006914) (Figure 2A), which may explain why KEGG analysis of this module enriched several cancer-related pathways (Figure 2B). In addition, pathways that respond to environmental stress, such as radiation response (GO: 0009314), were enriched. Next, we constructed a protein-protein interaction (PPI) network for the significantly differentially expressed genes in this module and identified six key genes: Adipoq, Scd1, Cyp2e1, Retn, Plin1, and Fabp4 (Figure 2C). WGCNA Analysis of Intestinal RNA Sequencing From Non-Treated (NT) and Irradiated (IR) Mice. (A) Sample Clustering. (B) Scale Independence. (C) Cluster Dendrogram. (D) Module-Trait Relationship Analysis. Effects of Radiation on Intestinal Tissue at Different Time Points after Irradiation. (A) GO Analysis of the Blue Module and (B) KEGG Analysis (C) PPI Diagram of Significantly Differentially Expressed Genes in the Blue Module. (D) GO Analysis of the brown Module and (E) KEGG Analysis of the Turquoise Module. (F) Upset Diagram of Differentially Expressed Exons at Different Time Points after Irradiation.

A brown module, which was enriched with numerous immune-related pathways, showed a significant negative correlation with the 3.5-day post-irradiation group (Figure 2D). In contrast, a turquoise module, enriched with DNA repair and neurodegenerative disease-related pathways, demonstrated a strong positive correlation with the 3.5-day post-irradiation group (Figure 2E). In the modules significantly associated with the 5-day group after irradiation, we also identified some pathways related to immunity and DNA repair, though the number of genes was notably smaller than that in the 3.5-day group.
Using the edgeR package, we analyzed alternative splicing across different time points after irradiation (Figure 2F). We observed that the expression of 47 and 4 exons was significantly higher or lower than pre-irradiation levels at the three time points post-irradiation. At least one exon of Lars2, Itpr1, Tctn1, Pappa, Pabpc4, Nol10, and Tnfrsf11b was significantly elevated at one or more time points after irradiation, with FDR values below 0.5. Furthermore, 97 and 17 exons were significantly elevated or reduced, respectively, at two or more time points compared to pre-irradiation.
Expression of DNA Damage-Related Genes in Intestinal Tissue 3.5 days After Irradiation
Given the observed changes in repair and immune genes 3.5 days post-irradiation and the role of Nrf2 in DNA repair, we irradiated wild-type mice and constructed Nrf2-KO mice, then sequenced intestinal tissues 3.5 days after irradiation (Figure 3). In this second sequencing, we identified a blue module enriched in DNA repair pathways and a brown module enriched in neurodegenerative disease pathways (Figure 4A and B). Considering the role of DNA repair in neurodegenerative diseases, we classified genes in these two modules as DNA repair-related genes.27,28 Subsequently, we constructed ceRNA networks using predicted target miRNAs for the repair-related genes with high differential expression (|logFc|>0.5) in both batches of samples 3.5 days post-irradiation. AK033546 and AK020274 established connections with 32 coding RNAs, and 68 predicted ceRNA-miRNA-codingRNA pathways were constructed (Figure 4C). A PPI network of protein products was generated from 68 highly expressed repair-related coding RNAs (|logFc|>0.8) 3.5 days post-irradiation in both sequencing results. MCODE analysis divided these genes into two clusters: one mainly nucleosomal components such as H3c7 (Hist1h3f), H3c8 (Hist1h3h), H4c11 (Hist4h4), H2bc12 (Hist1h2bk), H2bc4 (Hist1h2bc), and the other mostly ribosomal components such as Rpl7a, Rps5, Rpl14, and Rpsa (Figure 4D). WGCNA Analysis of Intestinal RNA Sequencing From Wild-type and Nrf2-KO Mice 3.5 days after Irradiation. (A) Sample Clustering. (B) Scale Independence. (C) Cluster Dendrogram. (D) Module-Trait Relationship Analysis. Regulatory Mechanisms of DNA Damage-Related Genes in Intestinal Tissue 3.5 days after Irradiation. KEGG Analysis of the Blue Module (A) Brown Module (B) in the Second Sequencing. DNA Repair-Related Genes with High Differential Expression Folds in Both Batches of Samples 3.5 days after Irradiation were Selected to Draw the ceRNA Network Sankey Diagram (C) and PPI Network Diagram (D).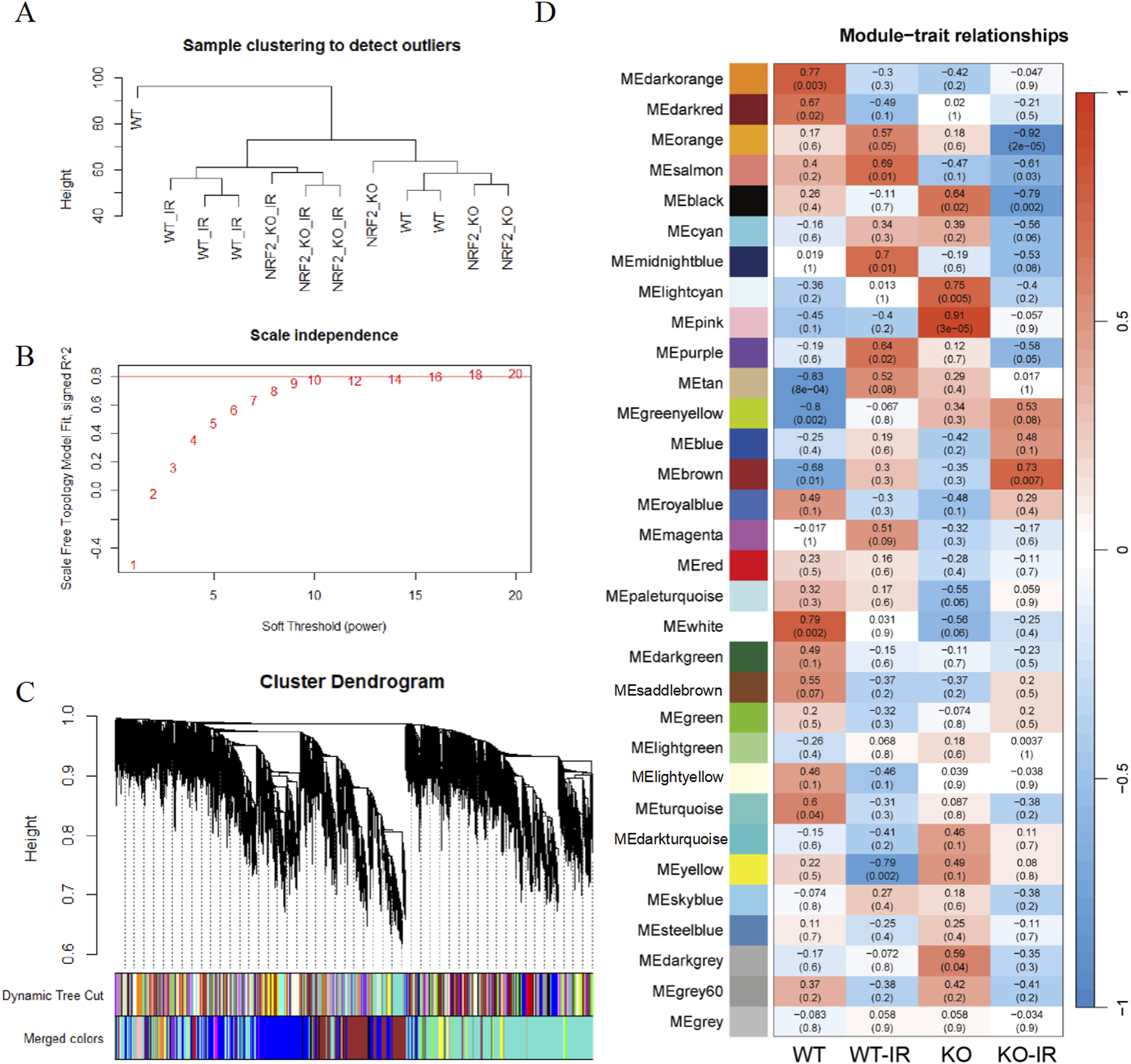

Expression of Immune-Related Genes in Intestinal Tissue 3.5 days After Irradiation
In the second sequencing, a turquoise module was enriched in immune-related genes (Figure 5A). Immune infiltration analysis showed significant reductions in T cells, CD4 memory, and B-derived cells after irradiation, while the levels of M0 and M1 cells increased significantly (Figure 5B and C). We verified the immune infiltration results and observed decreased infiltration of CD4 T cells and increased infiltration of CD8 T cells and macrophages at 3.5 and 5 days post-irradiation. Additionally, the infiltration level of DC cells decreased at 3.5 days after irradiation (Figure 5D). Module-immune cell correlation analysis indicated that the turquoise module was not significantly correlated with specific immune cell (Figure 5E). The intersection of the genes in the immune-related modules of the two sequencings was taken as a new gene set. After gene set variation analysis (GSVA) scoring, the new gene set showed significant positive correlations with T cells, CD4 memory, and B-derived cells, and significant negative correlations with M0 and M1 cells (Figure 5F). Regulatory Mechanisms of Immune-Related Genes in Intestinal Tissue 3.5 days After Irradiation. (A) KEGG Analysis of the Second Sequencing Turquoise Module. (B and C) Calculation of the Immune Infiltration Level of Samples Using CIBERSORT Package (B) and mMCPcounter Package (C) (*P < .05) (D) Ratios of CD4 T Cells and CD8 T Cells to the Intestinal Lamina Propria Cells and the Ratio of DC Cells and Macrophages to APC Cells at Different Time Points after irradiation (n = 3). (*P < .05, **P < .005, ***P < .0005, ****P < .0001) (E) Correlation Analysis Between the Module and the Level of Immune Cell Infiltration. (F) Correlation Analysis Between the Immune Gene Set after Merging Two Batches of Samples and the Level of Immune Cell Infiltration. (G and H) Select Immune-Related Genes With High Differential Expression Folds in the Two Batches of Samples 3.5 days after Irradiation, and Draw the ceRNA Network Sankey Diagram (G) and PPI Network Diagram (H).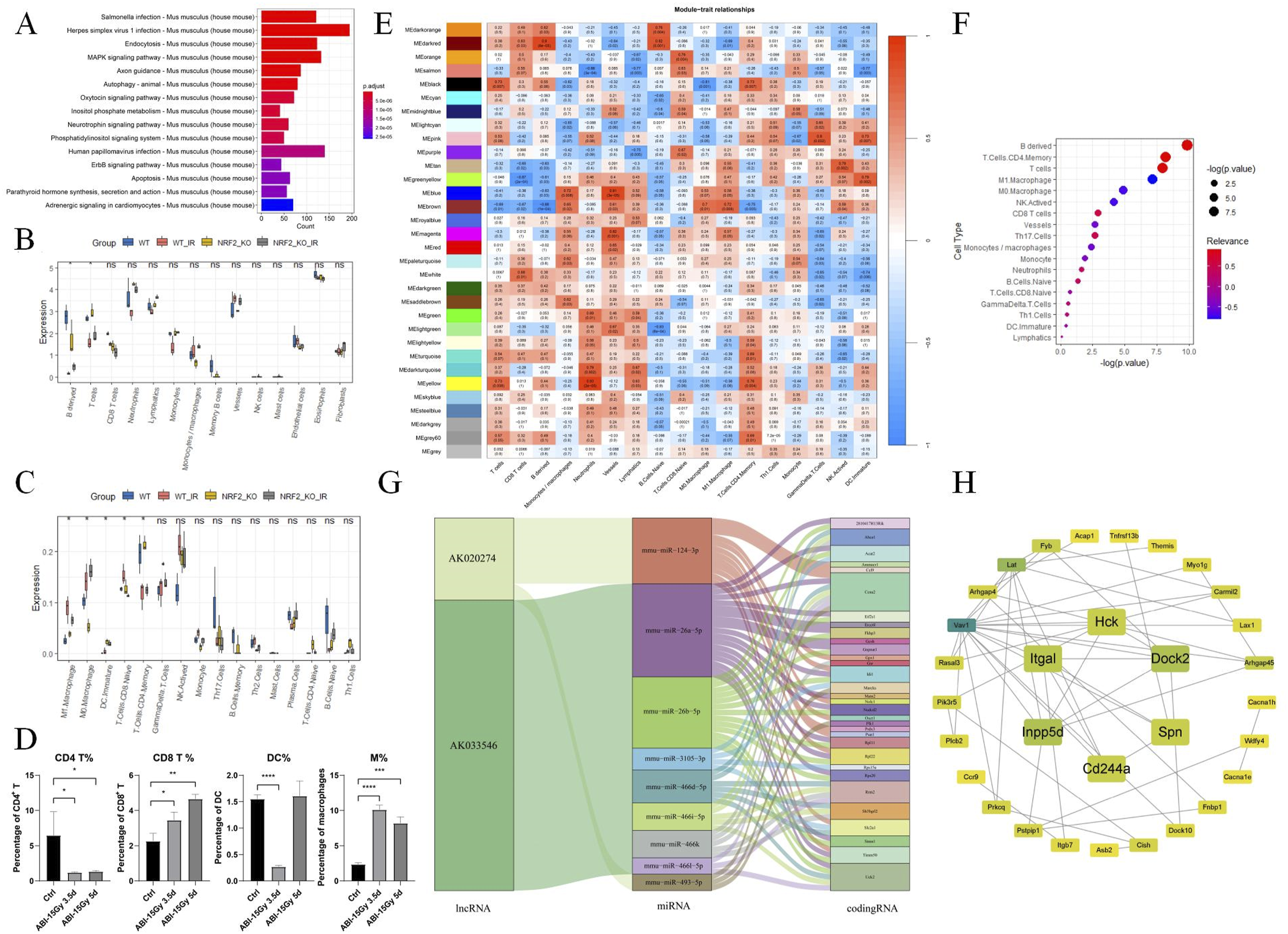
The blue and brown modules showed significant negative correlations with B-derived cells and significant positive correlations with monocytes/macrophages. We hypothesize that this may be due to the inclusion of some immune cell markers in these modules, though it remains unclear whether this correlation reflects a factual link between DNA repair and immune infiltration. Using predicted target miRNAs, we constructed ceRNA networks for immune-related genes with high differential expression (|logFc|>0.5) 3.5 days after irradiation from both sequencing results. AK143604, NR_001461, and uc009cfw.1 connected with 10 coding RNAs, resulting in 36 predicted ceRNA-miRNA-codingRNA pathways (Figure 5G). A subsequent PPI network for protein products of immune-related coding RNAs with high differential expression (|logFc|>0.5) 3.5 days post-irradiation in both sequencing results and MCODE identified Dock2, Hck, Inpp5d, Itgal, Spn, and Cd244a as hub genes (Figure 5H)).
Effect of Nrf2 Knockout on Radiation Effects in Intestinal Tissue 3.5 days After Irradiation
Given that Nrf2 functions as a common transcription factor, we verified the expression of Nrf2 target genes in the knockout mouse model. The salmon module correlated positively with WT and WT_IR groups and negatively with Nrf2_KO and Nrf2_KO_IR groups, suggesting that this module was enriched with Nrf2 target genes. In this module, the expression levels of 17 genes, including 1 lncRNA and 16 coding RNAs, were significantly higher in the WT and WT_IR groups than in the Nrf2_KO and Nrf2_KO_IR groups. However, only a few of these genes were related to radiation effects, including Nqo1, Gsta3, and Akr1b8.
In both circRNA and coding RNA, RNA methylation levels in the WT_IR group tended to be higher than in the WT group, and the methylation level in the Nrf2_KO group was significantly higher than that in the WT and Nrf2_KO_IR groups (Figure 6A-C). We speculate this result may be due to oxidative stress induced by both radiation and Nrf2 deficiency. However, we did not observe a significant correlation between RNA m5C levels and RNA expression. We analyzed the RNA expression levels of m5C-associated writer genes (Nsun family and Dnmt family), eraser genes (Tet family and Alkbh1), and reader genes (Alyref and Ybx1). The results showed that these genes were not significantly altered after irradiation. Effect of Nrf2 Knockout on Radiation Response in Intestinal Tissue 3.5 days After Irradiation. The Overall m5C Levels of (A) circRNAs (B) lncRNAs and (C) Coding RNAs in Intestinal Tissues of Wild-Type and Nrf2-KO Mice 3.5 days After Irradiation. The Expression Levels of Each Exon of the Pabpc4 Gene in the First Batch of Samples at 6 h (D), 3.5 days (E), and 5 days (F) after Irradiation Compared With the Non-Irradiated Group. In the Second Batch of Samples, the Differential Expression Levels of PABPC Exons in the WT-IR Group Compared With the WT Group (G) and the KO-IR Group (H) Compared With the KO Group. (I) Upset Plot of Differentially Expressed Exons in Different Groups.
We also performed alternative splicing analysis on the second sequencing results (Figure 6I). We found that two exons of two genes were significantly more expressed in the KO and KO_IR groups than in the WT and WT_IR groups, while five exons of five genes were significantly less expressed. This finding suggests that Nrf2 may play a role in alternative splicing of these genes, which is not affected by irradiation. 62 exons of 36 genes were significantly less expressed in the KO_IR group than in the other three groups. 15 genes, including Ganab, Fcho2, Specc1l, and Ofd1, had differentially expressed exons with an FDR value of less than 0.5 in at least one set of comparisons. In addition, 7 exons of 7 genes were significantly higher in the KO_IR group than in the other three groups. This may indicate the effect of Nrf2 knockout on alternative splicing of genes under irradiation conditions. In addition, we integrated the differential exons of the two sequencing data and found that only 6 exons such as exon 9 of Pabpc4 were differentially expressed before and after the irradiation (Figure 6). This shows that the conclusion on alternative splicing needs to be drawn through repeated cell experiments after bioinformatic analysis.
Changes in DNA Repair and Immune Infiltration in Intestinal Epithelium After Irradiation
To validate the robustness of our findings, we sought to determine whether publicly available datasets supported our conclusions. We identified dataset GSE123516, which used single-cell RNA sequencing to analyze both irradiated and non-irradiated intestinal epithelium.
23
Using the original authors’ classification methodology, we systematically categorized the cellular populations into seven distinct clusters. We further subdivided the lymphocytes into B cells, cytotoxic CD8 + T cells, and naive CD8 + T cell populations (Figure 7A, B and D). Quantitative analysis revealed a substantial reduction in CBC and PC populations following irradiation, with a complete ablation of B cells in the post-irradiation samples (Figure 7C and E). Despite the fact that our sequencing and validation were performed on the intestinal lamina propria, while the single-cell sequencing data were derived from the intestinal epithelium, the trend of reduced or absent B cells after irradiation remained consistent. Given the absence of lymph nodes within the intestinal epithelium, we hypothesize that the disappearance of B cells may reflect the depletion of B cells in the lamina propria following irradiation. Changes in DNA Repair and Immune Infiltration in Intestinal Epithelium After Irradiation. (A) Clustering of Intestinal Epithelial Cells. (B) Expression of Cell Markers Across Clusters. (C) Changes in the Number of Each Cell Cluster before and after Irradiation. (D) Further Clustering of Lymphocytes. (E) Changes in the Number of Different Lymphocyte Subtypes Before and After Irradiation. (F-H) Changes in DNA Repair Gene Scores for EC (F), EE (G), and GC (H) before and after Irradiation.
To further investigate the molecular changes associated with irradiation, we performed gene set variation analysis (GSVA) using the HALLMARK_DNA_REPAIR gene set (comprising 148 genes) obtained from the GSEA database (https://www.gsea-msigdb.org/gsea/msigdb). Our analysis showed that the GSVA scores for EE and EC significantly decreased after irradiation, whereas the GC scores significantly increased (Figure 7F-H). This finding contrasts with our sequencing results from the lamina propria, where DNA repair genes were significantly enriched after irradiation. This discrepancy highlights the distinct DNA repair capabilities between the intestinal epithelium and the lamina propria.
Discussion
The columnar epithelium of the gastrointestinal system is monolayered and is one of the fastest renewing tissues in the human body. 29 Renewal of the gastrointestinal mucosal epithelium after injury depends on gastrointestinal resident stem cells (SCs), which have the ability to self-renew and act as a repair system with unlimited division capacity. 29 Acute lesions of radiation-induced intestinal injury can occur during the irradiation period, mainly manifested by degeneration and desquamation of intestinal epithelial cells, dilation of capillaries in the intestinal wall, and congestion and edema of the intestinal wall. 30 We believe that the gene expression profile at 6 h after irradiation reflects the stress of intestinal tissue in the short period after irradiation, which is mainly manifested by inhibition of cell proliferation and motility and the activation of pathways related to environmental stress resistance. The key genes enriched in the relevant modules 6 h after irradiation are mainly lipid metabolism genes, and the high expression of these genes may be related to the processing of ROS. For example, Adipoq deficiency increases mitochondrial ROS, while FABP4 overexpression reduces radiation-induced ROS and γH2AX foci.31,32 Activation of the Nrf2/SCD1 pathway can prevent IR-induced neuronal ferroptosis. 33
Chromatin is composed of repetitively arranged nucleosomes, each consisting of 147 bp of DNA wrapped 1.75 times around the outside of the histone octamer. 34 When cells have difficulty repairing DNA damage caused by chemotherapy, cell cycle arrest occurs, leading to a decrease in histone transcription levels. 35 Li et al compared human cervical cancer cell lines with high and low expression of histone variants and found that the responses to DNA damage were different, suggesting that histone genes may have the function of protecting against DNA damage. 35 They also determined that HIST1H2BD, HIST1H2BJ, HIST1H2BH, HIST1H2AM and HIST1H4K can be used as prognostic factors for predicting the survival of cervical cancer patients. 35 In addition, Hao et al also found that the methylation level of the HIST1H4K gene decreased after irradiation, and the decrease in methylation usually also led to an increase in gene expression. 36 This is similar to what we observed. A large number of DNA repair pathways were enriched in the blue and brown modules of our group 3.5 days after irradiation, and the expression levels of 20 histone variant genes were significantly increased, including Hist1h2bj, Hist1h2bh, Hist1h4k, etc. screened by Li et al.
Eukaryotic organisms have 80S ribosomes, each of which consists of a small subunit (40S) and a large subunit (60S). 37 Approximately 80 different ribosomal proteins (r-proteins) can be found in eukaryotic ribosomes, and increasing evidence suggests that ribosomal proteins have functions beyond protein translation, including cell cycle, DNA repair, and maintenance of genome integrity.37,38 RPLP0 has been reported to bind to the RRM1 and RRM2 domains of NONO, a central paraspeckle component that has been shown to play pivotal roles in DSB repair. 39 RPLP0 deficiency may also be associated with PLAAT4-mediated growth inhibition and apoptosis. 40 In the absence of DNA damage, the DSB repair and response mediators RNF8 and BRCA1 are anchored to the nucleolus through the interaction of RNF8 with the ribosomal precursor protein RPSA. 41 Exposure of cells to γ-irradiation results in the relocalization of RNF8 and BRCA1 from the nucleolus to DNA damage foci. 41 Overexpression of RPL12 has been shown to increase the HR repair capacity of the Saccharomyces cerevisiae RS112 strain by two-fold. 42 In our study, we observed increased expression levels of RPLP0, RPSA, RPL12, and several other ribosomal protein genes 3.5 days after irradiation. This upregulation may be attributed to the heightened demand for protein synthesis during DNA damage repair. Alternatively, it could reflect the involvement of these ribosomal proteins in non-ribosomal functions, as suggested by previous studies.
The radiosensitivity of immune cells varies significantly based on their lineage, maturity, and activation state. 43 Natural killer (NK) cells, naive T cells, and B cells are highly sensitive to radiation, whereas effector T cells and NK/T cells demonstrate greater resistance. 43 Resident macrophages and circulating monocytes are sensors of tissue homeostasis disruption. 44 Upon tissue injury, activated macrophages release cytokines and chemokines to recruit neutrophils. 44 Monocytes in the bloodstream are recruited by neutrophils and differentiate into dendritic cells, resident macrophages (M2 type), or inflammatory macrophages (M1 type), depending on the tissue microenvironment. 44 Clinical studies have reported increased macrophage infiltration in rectal biopsies from patients with non-gastrointestinal pelvic cancer and prostate cancer following radiotherapy. 44 Similarly, experimental studies in rats and mice have observed elevated macrophage levels in intestinal tissue after irradiation.44,45 Our findings align with these observations, showing a significant increase in macrophage infiltration in intestinal tissue 3.5 days after irradiation, accompanied by a marked decrease in T cells and B cells. Analysis of key genes from the NCBI database (https://www.ncbi.nlm.nih.gov/gene) revealed that most were associated with the activation of T cells or B cells or the inhibition of myeloid cells. Supporting this, Shen et al demonstrated that combining a PIK3CD/G inhibitor with radiotherapy enhanced the anti-tumor immune effect of PD-1 blockade. 46 In addition, Vav1 was differentially expressed between radiation-resistant cells and radiation-sensitive cells, and GRAP2 responded specifically to low-dose radiation but not to high-dose radiation.47,48
Under normal redox conditions, NRF2 is located in the cell cytoplasm and is expressed at basal levels as a cell-protective enzyme. 49 When exposed to electrophilic chemicals or ROS, the kelch-like ECH-associated protein 1 (KEAP1) is inactivated. This inactivation allows newly synthesized NRF2 to escape KEAP1-mediated degradation, leading to its stabilization and accumulation within the cell. 49 NRF2 has been implicated in several DNA repair pathways, including direct repair, BER, HR, and NHEJ. To investigate the role of Nrf2 in DNA damage response, we used Nrf2 knockout mice to identify DNA damage effector genes regulated by Nrf2. 50 Through this approach, we identified three potential radiation-effect genes regulated by Nrf2: Akr1b8, Nqo1, and Gsta3. However, literature suggests that these genes likely mitigate DNA damage rather than enhance DNA repair.51-53 Notably, we did not detect differential expression of some common DNA repair genes regulated by Nrf2, such as 53BP1, MGMT, OGG1, FEN1, and RAD51D. We believe that this may be due to the selection of time points, differences between in vivo and in vitro experiments, and other variables.
Several limitations should be considered when interpreting the results of this study. First, the sample size was limited to three mice per experimental group due to budgetary constraints. While this number of biological replicates is generally considered sufficient to detect significant differences in gene expression patterns, a larger sample size could provide more robust statistical power. Second, the scope of our sequencing analysis was limited to mRNA, lncRNA and circRNA profiling, excluding miRNA sequencing. As a result, the construction of the ceRNA network relied on predicted interactions (lncRNA-miRNA, circRNA-miRNA, and miRNA-mRNA) from publicly available databases, which may not fully reflect the specific regulatory relationships in our experimental system.
Despite these limitations, our findings highlight the potential roles of histone variants and ribosomal proteins in DNA repair. Targeting these proteins with small molecules or other therapeutic interventions could enhance DNA repair capacity in normal tissues, thereby mitigating radiation-induced damage. Furthermore, by analyzing the expression profiles of radiation-effect genes and ceRNA networks in individual patients, it may be feasible to stratify patients according to their risk of developing gastrointestinal complications. This approach could pave the way for personalized treatment plans, optimizing radiation doses and schedules to minimize side effects, improve treatment tolerance, and ultimately enhance patient outcomes and well-being.
Conclusion
In summary, we identified the key differentially expressed genes in the small intestine 6 h after irradiation by RNA sequencing, focusing on the role of lipid metabolism in the intestinal stress response following radiation exposure. Our findings revealed enhanced DNA repair activity alongside impaired immune function 3.5 days after irradiation. Our study highlighted the role of histone variants and ribosomal proteins in DNA repair and mapped the ceRNA network for genes associated with both the DNA repair and immune modules. Additionally, we investigated the impact of Nrf2 knockout on DNA damage and identified Akr1b8, Gsta3, and Nqo1 as radiation-responsive genes regulated by Nrf2. We hope that our findings will contribute to the development of strategies for protecting normal intestinal tissue during radiotherapy, ultimately improving patient outcomes.
Footnotes
Statements and Declarations
Author Contributions
Jiale Li and Jinhan Wang conceptualized the study and drafted the manuscript. Jiale Li, Huanteng Zhang, Xiaoxiao Jia, Xumin Zong, and Shiqi Li analyzed the sequencing data, Jinhan Wang, Huanteng Zhang, Jiebing Guan, Xinran Lu, and Ningning He and Huijuan Song performed experiments. Kaihua Ji, Manman Zhang, Jianguo Li and Liu Qiang provided technical and funding support. Chang Xu supervised the study and revised the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (82273580 and 32171239), CAMS Innovation Fund for Medical Science (2021-I2M-1-042, 2023-I2M-2-008).
Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The original data has been uploaded to SRA, the data ID is PRJNA1187322 and will be made public on 2028-11-08.