
Abstract
Backgound
Traumatic brain injury (TBI) is a severe neurological disorders, which invloving complicated molecular mechanisms, such as endoplasmic reticulum (ER) stress and ferroptosis. , However, the mechanism underlying TBI remains unclear.
Objectives
The Objective was to determine the effect of VPA on ER stress and ferroptosis, and affirm the relationship between ER stress and ferroptosis. Methods: The expression levels of GRP78, ATF6, CHOP and GPX4 in brain tissues were detected via western blot, histological staining, and immunofluorescence. The effect of VPA on ER stress and ferroptosis on OS cellswas evaluated in vitro and in vivo.
Results
In our study, we found that VPA suppressed ER stress after TBI by inhibiting the GRP78-ATF6-CHOP signaling pathway, which ameliorated ferroptosis by reversing the reduction of the ferroptosis protein GPX4. Furthermore, tissue defects, bleeding, and iron accumulation also reduced. Moreover, 4-phenylbutyric acid was used to further confirm our assumption.
Conclusion
VPA plays a neuroprotective role by inhibiting ER stress levels and subsequently inhibiting ferroptosis.
Introduction
Traumatic brain injury (TBI) is a severe public health problem associated with high disability. 1 TBI is often caused by mechanical injury due to a sudden impact, leading to multiple secondary injury involving encephaledema, inflammatory response, ionic homeostasis, and oxidative stress.2-4 These physiological processes are closely linked to and programmed cell death such as apoptosis, pyroptosis, and ferroptosis.5,6 Although numerous studies have focused on TBI during the past decades, the mechanis ms underlying the onset of TBI remain unclear. There is also an urgent need to explore effective drugs and therapeutic strategies for TBI.
The endoplasmic reticulum (ER) is an critical organelle in maintaining normal physiological functions, such as protein synthesis, modification, and transport. ER stress is a critical component of secondary injury following TBI, leading to the accumulation of misfolded proteins in the ER, and finally contributing to ER dysfunction. 7 ER stress involves 3 different signaling pathways, including PERK signal pathway, ATF6 signal pathway and IRE1 signal pathway, which are associated with GRP78. 8 Upregulation of GRP78 expression induces changes in ER stress–related proteins such as ATF6 and CHOP.8,9 Sustained abnormal ER stress contributes to neuronal dysfunction and death. 10 Nevertheless, ER stress inhibition improves TBI-related pathological process and ameliorates neurological deficit after TBI. 11 However, the role of ER stress in TBI is not well understood, and the underlying regulatory mechanisms require further exploration.
Ferroptosis, a novel form of programmed cell death, is characterized by abnormalities in the metabolism of iron, inactivation or decreased expression of ferroptosis-related proteins, and accumulation of lipid peroxide. 12 Iron dyshomeostasis is a strong inducer of secondary damage after TBI. 13 Inhibition of ferroptosis induction after TBI alleviated neuronal injury and neurodegeneration caused by TBI. 14 An interaction exists between ER stress and ferroptosis. Erastin-induced ER stress upregulated ATF4 expression, which increases SLC7A11 levels to limit ferroptosis. 15 Furthermore, inhibition of ER stress alleviated ferroptosis induced by acute cadmium toxicity and improves cell viability. 15 However, the relationship between ER stress and ferroptosis after TBI remains unclear.
Valproic acid (VPA), 1 of the safest mood stabilizers, has shown neuroprotective effects in multiple animal models of neurological diseases.16,17 VPA treatment significantly inhibits ER stress and apoptosis induced by seizures. 18 Furthermore, GRP78, the critical protein of ER stress, is a VPA-regulated gene in the rat cerebral cortex. 19 VPA also exerts a positive effect in neuroprotection and anti-apoptosis after TBI.20,21 Nevertheless, the effect of VPA on ER stress after TBI remains ambiguous. Considering the enhanced levels of ER stress after TBI, we hypothesized that VPA reduces ferroptosis and ameliorates TBI by inhibiting ER stress after TBI and that ER stress may be a potential target for TBI therapy.
In this study, we investigated the relationship between ER stress and ferroptosis by combining with the ER stress inhibitor 4-phenylbutyric acid (4-PBA) and explored the role of VPA in ER stress and ferroptosis after TBI.
Materials and Methods
Animals and Ethics Statement
Adult C57BL/6 8-week-year-old male mice weighing 20-25 g were used in current study. All animals were housed under a 12 h light/dark cycle at 21-23°C and received free access to food and water. All the experimental procedures were approved in advance by the Laboratory Animal Ethics Committee of WenZhou Medical University (Approval number: wydw2023-0571), and conducted in strict accordance with the National Institutes of Health Guidelines for the Care and Use of Experimental Animals. All the mice were divided into 4 groups randomly, including the sham group, the TBI group, the VPA group (300 mg/kg, I.P.) and the 4-PBA group (100 mg/kg, I.P.). The detailed protocol was shown as in Figures 1(A) and (B). Experimental scheme for VPA administration after TBI. (A) Time diagram of brain injury, drug administration, and experimental detection in C57BL/6 male mice. VPA, valproic acid; 4-PBA, 4-Phenylbutyric Acid; TBI, traumatic brain injury. (B) Schematic diagram of TBI modeling operation.
Cell Culture and Drug Delivering
HT-22 cells and SH-SY5Y cells were purchased the Cell Bank of the Type Culture Collection of the Chinese Academy of Sciences, Shanghai Institute of Cell Biology, Chinese Academy of Sciences. All the cells has been certified and tested for mycoplasma contamination and no mycoplasma was detected. All the cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum and 1% penicillin-streptomycin solution in an incubator at 37°C and 5% CO2. To clear the effect of VPA, we induced ER stress by stimulating cell with TBHP (300 μm) for 2 h, and then VPA (3 mM) was delivered as a therapeutic drug. To further understand the effect of ER stress, cells were pretreated with 4-PBA (1 mM) for 1 h.
Procedure for TBI Model
All male mice were anesthetized with isoflurane via anethesia instrument. After the induction box was filled with gaseous isoflurane at a concentration of 4%, the animals were put into the induction box for 2 min, making ensure the animals were anesthetized quickly, with less fear and pain. The mice were then placed in the brain stereotaxis, and remained stupefacient by being received isoflurane at a concentration of 1.5% via a face mask. An incision was made in the middle of the scalp to expose the skull with a hand-held cranial drill, followed by controlled cortical impact (CCI) with a controlled impactor. The specific setting of the impact was shown as follows: the speed of impact was set at 4 m/s, penetration depth was set at 1.0 mm, and the impactor dwell time was 600 ms. The scalp was sutured immediately when procedure was finished. During the surgery, a heating pad was used to maintain the animals body temperature until they wake up from anesthesia. Sham group accept the same operation except impact. All effort had been made to reduce the number of animals used as well as their suffering.
Drug Administration
In this study, Valproic acid and 4-PBA were used as therapeutic drug. VPA was dissolved as 150 mg/ml, and 4-PBA was dissolved as 100 mg/ml. The mice were treated with 300 mg/kg of VPA by intraperitoneal injection 30 min after TBI, and then once a day for 3 days. 100 mg/kg 4-Phenylbutyric Acid was delivered 3 days before TBI to inhibit ER stress.
Sample Preparation
We mainly focus on the molecular mechanisms at 3 days after TBI. On d3, the mice were anesthetized by an intraperitoneal injection of 1% pentobarbital sodium (40 mg/kg) and then were sacrificed by achieving bloodletting death with a perfusion needle. For immunohistochemical staining, HE staining, Nissl staining and Prussian Blue staining, the brain was taken out, fixed with 4% paraformaldehyde at 4°C for 48 h, embedded into paraffin and cut into 5 μm sections. The rest of the mice were used for Western Blotting. The brain tissues were removed and stored in liquid nitrogen. Then the fresh brain segment was placed in the RIPA lysis buffer including 1% PMSF, and then ground in a multi-sample tissue homogenizer at 4°C for 90 s to extract tissue protein. Subsequently, the supernatant was collected after centrifuging at 12 000 r/min at 4°C for 1 min. All the experiments were performed in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Wesrern Blotting Analysis
The BCA reagent was used to quantify the concentration of each protein supernatant from tissues or cells. The protein was separated in a 10% or 12.5% SDS-PAGE gels, then transferred to PVDF membrane. The membrane was blocked with 5% skim milk, which dissolved in a tris-buffer solution for 1 h. The membrane was cut horizontally into different bands according to protein marker for further detection, and then these bands were incubated overnight at 4°C with primary antibody according to molecular weight including β-actin (1:1000), ATF6 (1:1000), GRP78 (1:1000), CHOP (1:1000), PDI (1:1000), GPX4 (1:1000), and FTH (1:1000). On the next day, the membranes were incubated with the corresponding secondary antibody at RT for 1 h. The signal of protein was detected by Chemiluminescence imaging system, and the relative density of the band was quantified by ImageJ software.
Hematoxylin and Eosin (HE) Staining, Nissl Staining and Prussian Blue Iron Staining
The sections of the brain were immersed in xylene twice for 20 min at RT, then were incubated in a gradient ethanol solution (100, 90, 80 and 70%) and pure water for 10 min. Subsequent operation observed the manufacture’s protocol. Briefly, for H&E staining, the sections were immersed in hematoxylin for 2 min eosin for 2 min. For Nissl staining, the sections were stained with cresyl violet 2 min, and nissl differentiation solution 30 s. For Prussian Blue Iron Staining, the sections were incubated in operating fluid at 37°C for 1 h, and then immersed in nuclear fast red for 5 s. When the above operations were finished, the sections were immediately dehydrated by been immersing in following solutions: twice in 95% alcohol for 2 s, once in 100% alcohol for 2 s, once in 100% alcohol for 1 min, and twice in xylene for 1 min, follow by sealing with neutral gum and being imaged with optical microscope.
Immunofluorescence Staining
The prepared tissue sections and cells were blocked with 5% BSA for 1 h at 37°C and then incubated with primary antibody overnight at 4°C. The primary antibodies were shown below: GPX4 (1:500, Abcam, United States) and CHOP (1:200, Proteintech, United States). On the next day, the section and cells were incubated with Alexa Flour 488 or Alexa Flour 647 donkey anti-rabbit/anti-mouse secondary antibody for 1 h at 37°C. We use DAPI as a unclear staining and Leica microscope to detect the intensity of fluorescence signal. Every sections from different group were captured under the same laser intensity.
Statistical Analysis
All the statistical analysis was performed using Graphpad Prism 8 software. Student’s t-test was used to determine significant differences between 2 groups. One-way ANOVA with Tukey’s Multiple comparison test was performed when 2 or more group are compared. And two-way ANOVA analysis, followed by Sidak’s or Tukey’s Multiple comparison test, was used when these are 2 variables. P < 0.05 was considered statistically significant.
Results
VPA Ameliorates Tissue Loss and Iron Accumulation Induced by TBI
Our previous study revealed that VPA exerts a neuroprotective effect on TBI; thus, promoting neurological functional recovery. However, the effect of VPA on mechanistic changes in the acute phase of TBI remains unclear. To determine the effect of VPA on the pathological process of TBI during the acute phase, hematoxylin and eosin (H&E) staining and Nissl staining were performed. The TBI group showed severe tissue loss in the cerebral cortex at 3 days after TBI when compared with the sham group (Figure 2(A)). However, VPA treatment decreased tissue loss and reduced vacuolation in the cerebral cortex. Similar to the results of H&E staining, Nissl staining showed that VPA treatment reduced neuronal apoptosis in the cerebral cortex. Given that ER stress plays an important role in functional recovery after TBI, we investigated whether downregulation of ER stress alleviates TBI-induced tissue damage by administering the ER stress inhibitor 4-PBA. The results of the staining assay showed that tissue loss and neuronal damage in the cerebral cortex improved after 4-PBA treatment, when compared with those in the TBI group (Figure 2(A), (B) and (D)). Additionally, several studies indicated that the relative levels of ferroptosis were anabatic after TBI. Prussian blue iron staining was used to observe iron accumulation in different groups, and the results showed that VPA treatment reduced injury due to iron accumulation (Figure 2(C)). Interestingly, the 4-PBA group showed remission of iron accumulation, which suggested that some association existed between ER stress and ferroptosis. However, no studies have described the detailed relationship between ferroptosis and ER stress, which needs further exploration. VPA treatment alleviates nerve damage and inhibits iron accumulation during the acute phase of TBI. (A) Representative images of Hematoxylin-Eosin (HE) staining of the brain tissues at 3 dpi (10 × ), scale bar = 100 μm. (B) Representative images of Nissl staining of the brain tissues (10 × ) at 3 dpi, scale bar = 100 μm. (C) Representative images of Prussian Blue Iron Staining of the brain tissues (40 × ); arrows indicate the occurrence of iron-accumulation at 3 dpi, scale bar = 50 μm. (D) Quantitative analysis of the number of nissl bodies in (B), n = 3;TBI group vs VPA group: ***P < 0.001, TBI group vs 4-PBA group: **P < 0.01.
Relative Activation of ER Stress and Upregulated Expression of Ferroptosis-Related Proteins after TBI
To further investigate the relationship between ER stress and ferroptosis levels, we first determined changes in the expression levels of ER stress–related proteins and ferroptosis-related proteins. The expression level of the GRP78 protein, an ER stress marker, was substantially increased (Figures 3(A) and (B)). GPX4, the key protein associated with ferroptosis, showed a contrasting tendency (Figures 3(D) and (E)), which indicated that the levels of ER stress and ferroptosis were significantly enhanced on day 3 after TBI onset. Additionally, changes in the levels of ER stress and ferroptosis were observed by immunofluorescence staining. The results showed that the number of CHOP-positive cells in the TBI group was significantly higher than that in the sham group, whereas the number of GPX4-positive cells in the TBI group was significantly lower than that in the sham group (Figures 3(C) and (F)). Given that both ER stress and ferroptosis play important roles in the pathological process and functional recovery after TBI, we hypothesized that inhibiting ER stress would regulate ferroptosis. Relative activation of ER stress and upregulated expression of ferroptosis-related proteins after TBI. (A) Representative western blotting images of GRP78 expression level at 3 dpi. (B) Quantification of GRP78 expression in (A), n = 3, sham group vs TBI group: **P < 0.01. (C) Representative immunofluorescence staining images of CHOP (green) expression level at 3 dpi, scale bar = 50 μm or 200 μm. (D) Representative western blotting images of GPX4 expression level at 3 dpi. (E) Quantification of GPX4 expression in (D), n = 3, sham group vs TBI group: ****P < 0.0001. (F) Representative immunofluorescence staining images of GRP78 (green) expression level at 3 dpi, scale bar = 50 μm or 200 μm.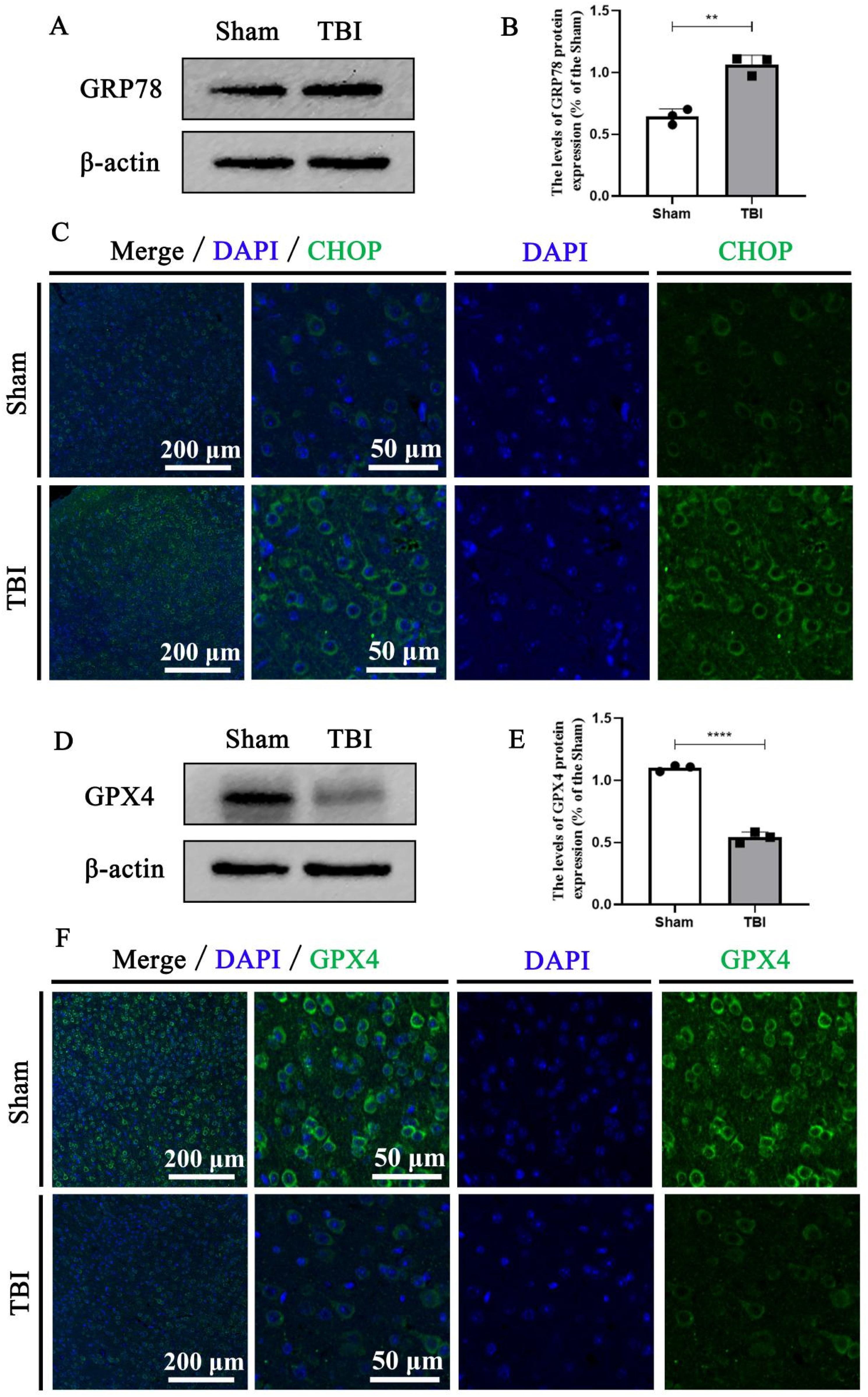
VPA Attenuated the Activation of ER Stress after TBI by Downregulating the Expression of GRP78, ATF6, and CHOP Proteins
VPA promoted neurological function recovery. However, the effect of VPA on ER stress during acute TBI remains unclear. Recently, inhibition of ER stress was found to weaken the secondary effect after TBI, such as by reducing neuronal apoptosis and ameliorating neuroinflammation.
22
Thus, ER stress might be a potential target for TBI treatment. To determine the relationship between VPA and ER stress, western blotting and immunofluorescence staining were performed to observe the effect of VPA on the expression of proteins related to the ER stress signaling pathway. The expression of ER stress–related proteins such as GRP78, ATF6, and CHOP was markedly anabatic on day 3 after TBI, which was reversed by VPA treatment (Figures 4(A) to (D)). The results of immunofluorescence staining assay also indicated that VPA delivery decreased the upregulated expression of CHOP-positive cells caused by TBI (Figure 4(E)). Next, the effect of VPA on ER stress was confirmed in the 4-PBA group. The results demonstrated that 4-PBA administration effectively inhibited protein expression levels of GRP78, ATF6, and CHOP (Figures 4(A) to (D)). These findings revealed that VPA attenuated ER stress levels during the acute phase of TBI, involving changes in the expression of GRP78, ATF6, and CHOP. VPA attenuated the activation of ER stress after TBI by downregulating the expression of GRP78, ATF6, and CHOP proteins. (A) Representative western blotting images of ATF6, GRP78 and CHOP expression level in the 3 days post-TBI. (B) Quantification of ATF6 expression in (A), n = 3, sham group vs TBI group: ***P < 0.001; TBI group vs VPA group:*P < 0.05; TBI group vs 4-PBA group: **P < 0.01. (C) Quantification of GRP78 expression in (A), n = 4, sham group vs TBI group: ***P < 0.001; TBI group vs VPA group: *P < 0.05; TBI group vs 4-PBA group: **P < 0.01. (D) Quantification of CHOP expression in (A), n = 3, sham group vs TBI group: ***P < 0.001; TBI group vs VPA group: ***P < 0.001; TBI group vs 4-PBA group: ****P < 0.0001. (E) Representative immunofluorescence staining images of CHOP (green) expression level in the 3 days post-TBI, scale bar = 50 μm or 200 μm.
VPA Inhibited the Relative Activation of Ferroptosis after TBI
Ferroptosis is closely associated with the development of various neurodegenerative diseases such as Parkinson’s disease, Alzheimer’s disease, and amyotrophic lateral sclerosis.
23
Accumulating evidence suggests that ferroptosis exerts a critical effect in TBI, which is a crucial molecular event after TBI.6,13 Although ferroptosis inhibition after TBI can ameliorate the recovery of neurological function in animals, no study has specifically elaborated on the molecular mechanism underlying ferroptosis after TBI.14,24 Because of the decline in iron accumulation after VPA or 4-PBA treatment (Figure 1(C)), we assume that VPA or 4-PBA probably inhibited ferroptosis levels. Thus, we detected GPX4 expression, a key protein involved in the process of ferroptosis that converts lipid hydrogen peroxide into nontoxic lipid alcohols. VPA treatment significantly rescued GPX4 expression, which showed a sharp decline in the TBI group (Figures 5(A) and (B)). The results of immunofluorescence staining displayed the same tendency (Figure 5(C)). The number of GPX4-positive cells was remarkably descended, and VPA reversed the downregulated expression of GPX4 after TBI. Furthermore, the relationship between ER stress and ferroptosis was explored using 4-PBA, a classical inhibitor of ER stress. Interestingly, the protein expression level of GPX4 was substantially downregulated in the 4-PBA group relative to that in the TBI group (Figures 5(A) and (B)). Next, immunofluorescence staining assay also displayed that 4-PBA treatment reversed the reduction in the number of GPX4-positive cells (Figure 5(C)). Based on these findings, we concluded that VPA-mediated protection may be exerted through the inhibition of the activity levels of ER stress and ferroptosis, and the decrease in ferroptosis may be related to the inhibition of ER stress. VPA inhibited the relative activation of ferroptosis after TBI. (A) Representative western blotting images of GPX4 expression level in the 3 days post-TBI. (B) Quantification of GPX4 expression in (A), n = 5, sham group vs TBI group: ****P < 0.0001; TBI group vs VPA group: ***P < 0.001; TBI group vs 4-PBA group: *P < 0.05. (C) Representative immunofluorescence staining images of GPX4 (green) expression level in the 3 days post-TBI, scale bar = 50 μm or 200 μm.
VPA Suppressed TBHP-Induced ER Stress and Ferroptosis Levels in SH-SY5Y Cells
We further investigated the effect of VPA treatment on ER stress and ferroptosis in vitro using SH-SY5Y cells. Considering that the production of reactive oxygen species (ROS) is a major cause of secondary damage after TBI, SH-SY5Y cells were treated with tert-butyl hydroperoxide (TBHP), a damage-causing peroxide, to simulate the in vitro model of TBI. Western blotting analysis was performed to evaluate the GRP78-ATF6-CHOP signaling pathway (Figures 6(A) to (D)). The results revealed that ER stress–related proteins (GRP78, ATF6, and CHOP) had sharply upregulated expression after TBHP treatment, which was rescued after VPA administration. The results of immunofluorescence staining assay further confirmed our conclusion (Figure 6(F)). We also assessed the protein expression of GPX4. As predicted, GPX4 expression levels decreased in the TBHP group, implying enhanced ferroptosis (Figure 6(A), (E) and (G)). However, VPA treatment restored GPX4 expression levels. These conclusions further confirmed our previous speculations that VPA inhibits ER stress and ferroptosis levels after TBI. Subsequently, we suspected that enhanced ER stress might contribute to the reinforcement of ferroptosis. Inhibition of ER stress levels may attenuate ferroptosis levels, thereby promoting neurological recovery after TBI. VPA suppressed TBHP-induced ER stress and ferroptosis levels in SH-SY5Y cells. (A) Representative western blotting images of GRP78, ATF6, CHOP and GPX4 expression level in the SH-SY5Y cell. (B) Quantification of GRP78 expression in (A), n = 4, con group vs TBHP group: ***P < 0.001; TBHP group vs VPA group: *P < 0.05. (C) Quantification of ATF6 expression in (A), n = 3, con group vs TBHP group: **P < 0.01; TBHP group vs VPA group: **P < 0.01. (D) Quantification of CHOP expression in (A), n = 3, con group vs TBHP group: **P < 0.01; TBHP group vs VPA group:*P < 0.01. (E) Quantification of GPX4 expression in (A), n = 3, con group vs TBHP group: ***P < 0.001; TBHP group vs VPA group: **P < 0.01. (F) Representative immunofluorescence staining images of CHOP (green) expression level in SH-SY5Y cell, scale bar = 60 μm. (G) Representative immunofluorescence staining images of GPX4 (green) expression level in the HT-22 cell, scale bar = 60 μm.
Inhibition of ER Stress Activity Levels Reserved the Upregulated Expression of Ferroptosis Induced by Erastin in HT-22 Cell Lines
To further explore the relationship between ER stress and ferroptosis, we induced the ferroptosis model of HT-22 using erastin, one of the most commonly used ferroptosis inducer. VPA and 4-PBA were then administered, and the relationship between ER stress and ferroptosis was analyzed by western blotting and immunofluorescence staining assays. The protein expression level of GPX4 was significantly decreased in the erastin group (Figure 7(A) to (C)). However, as the ER stress levels were inhibited, the protein expression of GPX4 in the 4-PBA and VPA groups was restored. All these results suggest that inhibition of ER stress levels can rescue the downregulated expression of the GPX4 protein and then inhibit ferric death activity Figure 8. Inhibition of ER stress activity levels reserved the upregulated expression of ferroptosis induced by erastin in HT-22 cell lines. (A) Representative western blotting images of GPX4 expression level in the HT-22 cell. (B) Quantification of GPX4 expression in (A), n = 3, con group vs Erastin group: **P < 0.01; Erastin group vs VPA group: *P < 0.05; Erastin group vs 4-PBA group: *P < 0.05. (C) Representative immunofluorescence staining images of GPX4 (green) expression level in the HT-22 cell, scale bar = 60 μm. Graphical abstract of the mechanisms of VPA on ER stress and ferroptosis after TBI.

Discussion
Owing to complex secondary injuries after TBI, such as inflammation, apoptosis, and ER stress, TBI is currently 1 of the most difficult public health problems worldwide.3,25,26 In the past few decades, great progress has been made in TBI research. However, no effective therapy is available in the clinical setting. Therefore, it is important to explore new targets of TBI, which may provide further ideas for the development of new therapies. In this study, we found that mice that received an intraperitoneal injection of VPA for 3 days after TBI had reduced tissue loss and improved iron accumulation in the cerebral cortex, which reflected the neuroprotective effect of VPA on TBI. However, the underlying molecular mechanisms remain unclear.
VPA is widely used as a mood stabilizer to treat psychiatric disorders such as epilepsy, bipolar disorder, and migraine, which exerts its effects by affecting inhibitory synapses, blocking various voltage-gated channels, and inhibiting histone deacetylase activity.27-30 Increasing evidence has shown that VPA plays a neuroprotective role in other diseases related to the central nervous system by affecting other physiological events, including inflammatory responses, autophagy flow, apoptosis, oxidative stress, and ER stress.20,31-33 Biesterveld et al 34 indicated that post-traumatic VPA treatment significantly reduced the area of brain injury after TBI and revealed that VPA alters various molecular events after TBI, including calcium signaling pathways, mitochondrial metabolism, and biosynthesis. However, the specific mechanism of action of VPA remains unclear. Therefore, determining the mechanism underlying the neuroprotective effect of VPA after TBI offers a new direction for the development of therapeutic strategies for TBI.
ER stress participates in the pathological process of TBI, which suggests that the regulation of ER stress after TBI may be conducive to ensure VPA as a therapeutic strategy of TBI.10,11 Xie et al 35 reported that moderate ER stress may alleviate cell injury and apoptosis. However, excessive ER stress induced by traumatic injury or neurodegenerative diseases contributes to serious dysfunction of neurological function. 36 Luo et al 37 demonstrated that selective inhibition of the activity of GRP78 significantly suppressed ER stress and inflammatory responses. In our study, we confirmed that ER stress was involved in the treatment of TBI with VPA by comparison with the mice injected with 4-PBA intraperitoneally. We also observed that the protein expression of GRP78, ATF6, and CHOP significantly increased 3 days after TBI onset, which was reversed after VPA treatment (Figure 4(A)). Treatment with the ER stress inhibitor 4-PBA also decreased tissue loss 3 days after TBI onset (Figures 1(A) and (B)). Thus, VPA treatment ameliorates tissue structural damage and reserves neuronal death by inhibiting ER stress.
Ferroptosis is known as iron-dependent programmed cell death, characterized by severe lipid peroxidation induced by ROS and iron overload.12,38 Recently, increasing evidence affirmed that ferroptosis was involved in the development of various neurological disorders such as stroke, spinal cord injury, and TBI.39-41 Excessive ferroptosis induced by TBI resulted in cognitive deficits, neurodegenerative disorders, and post-trauma epilepsy.42-44 By contrast, diminishing ferroptosis activity improves functional recovery and prognosis after TBI. 45 A detailed understanding of the regulatory mechanism of ferroptosis will offer a new strategy for TBI treatment, whereas the exact mechanisms remain undetermined. Zhang et al 46 revealed that ER stress inhibition upregulated the expression of peroxisome proliferator–activated receptor-λ, a critical gene that regulates lipid peroxide accumulation, leading to ferroptosis and subsequently inhibiting acrolein-induced ferroptosis. Interestingly, our study also revealed that the expression of GPX4, the crucial protein of ferroptosis, significantly decreased after the inhibition of TBI-induced ER stress, which led to the speculation that an unclear connection may exist between ER stress and ferroptosis in the pathological process of TBI. Furthermore, we investigated whether the enhanced ferroptosis was related to the upregulated expression of ER stress. We tested the effect of VPA on ferroptosis in the cerebral cortex after TBI. GPX4 plays a crucial role in iron death, and the decline in the expression of GPX4 is 1 of the main reasons for the enhanced ferroptosis activity. Therefore, we evaluated GPX4 protein expression in the cerebral cortex after VPA treatment. The results showed that GPX4 protein expression decreased significantly after TBI, while VPA could restore GPX4 expression (Figures 5(A) and (B)). To confirm whether the downregulation of ferroptosis is related to ER stress, we monitored iron accumulation in the cerebral cortex after 4-PBA administration. As expected, ER stress inhibition effectively alleviated iron accumulation, improved histological morphology, and decreased neuronal death (Figure 1(C)). Subsequently, western blotting and immunofluorescence staining assays were performed. Moreover, 4-PBA inhibited the expression of ER stress–related proteins (GRP78, ATF6, and CHOP) and restored the high expression level of GPX4 (Figures 4(A) and (B)). The ferroptosis inducer erastin was used to induce an in vitro ferroptosis model in HT-22 cells. The results were the same as those observed after treatment with VPA or 4-PBA. Taken together, all these results indicated that VPA suppressed ferroptosis by inhibiting ER stress and restoring GPX4 expression level, thereby exerting a neuroprotective effect on TBI. However, whether VPA directly inhibits ferroptosis by downregulating ER stress requires further research.
In summary, our study confirmed that VPA administration downregulated ER stress after TBI onset by inhibiting the GRP78-ATF6-CHOP signaling pathway and reduced tissue defects, bleeding, and iron accumulation. We also found that specific inhibition of ER stress activity levels ameliorated ferroptosis, thus, reversing the reduction of GPX4 levels, a key protein for ferroptosis. Although there have been extensive advances in TBI treatment in the past few decades, there is currently no effective treatment. It is of great significance to explore new therapies for TBI. Our conclusion supports that VPA administration ameliorates enhanced TBI-induced ER stress and restores GPX4 expression, thereby inhibiting ferroptosis levels, decreasing tissue loss, and reducing neuronal death after TBI, indicating that VPA may be a potential drug for TBI treatment.
Footnotes
Acknowledgments
The authors would like to thank the School of Pharmaceutical Sciences (Wenzhou Medical University) for technical assistance. Some illustrations in this study were created using BioRender.com
Author Contributions
Jie Chen: Carried out laboratory research, collected samples. Lei Li: Designed the experiment, carried out laboratory research and theoretical research, wrote draft of manuscript. Lei Huang: Revised the manuscript. Chengyu Zhao: Prepared the TBI mice model and collected samples. Zhanwei Ruan: Designed the experiment, carried out theoretical research, revised manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Natural Science Foundation of Zhejiang Province (LY23H060004), the Medical and Health Research Project of Zhejiang Province (2021KY1082), Wenzhou Science and Technology Innovation Project (ZY2020026) and Wenzhou Medical Association Scientific Research Project (202303KZ1), Natural Science Foundation of Ningbo (2021J032, 2022J044).