
Abstract
The health of radiation workers has always been our focus. Epidemiological investigation shows that long-term exposure to low-dose ionizing radiation can affect human health, especially cancer and cardiovascular disease, and there are many studies on it. However, up to now, there have been few reports on the research of blood and biological samples from radiation workers. In this study, radiation workers and healthy control groups were strictly screened, and the transcriptome of mRNA and circRNA was sequenced by extracting their peripheral venous blood. At the same time, appropriate data sets were selected in the GEO database for bioinformatics analysis, and circRNA-miRNA-mRNA network was constructed. We identified 9 different circular ribonucleic acids, 3 tiny ribonucleic acids, and 2 central genes (NOD 2 and IRF 7). These differentially expressed genes and non-coding RNA are closely related to ionizing radiation damage, and play an important role as biological markers. In conclusion, this study may provide new insights into the role of the circRNA-miRNA-mRNA regulatory network in the health of radiation workers, and provides a new strategy for the future study of radiation biology.
Keywords
Introduction
The peripheral blood examination of radiation workers is an indispensable examination in the current occupational health examination. By analyzing a series of indicators such as blood routine, blood biochemical changes, chromosome aberration, and micronucleus rate (quoted), it can be judged whether the workers suffer from radiation diseases and whether they are suitable for the current radiation posts.1,2 However, in most cases, long-term exposure to low-dose ionizing radiation may be within normal range of peripheral blood examination, which may lead to neglect of potential disease risk. 3 The risks of disease caused by long-term low-dose ionizing radiation usually have a long incubation period. If the changes of peripheral blood transcriptomics are used to judge the exposure level of workers, the possibility of continuing to engage in radioactive work and the possibility of suffering from certain diseases, it will be better to monitor the health status of radiation work.3,4 In recent years, with the development of transcriptome technology, it is possible to detect non-coding RNA and mRNA in blood samples. 5 Therefore, in order to better protect the health of radiation workers, it is necessary to systematically analyze the changes of transcriptome of blood samples of radiation workers, hoping to find possible potential markers and better determine their health status.
CircRNA is a specific endogenous non-coding RNA, which has evolved and preserved in eukaryotic species and is widely expressed in high abundance human cells. The closed-loop structure makes the cyclic ribonucleic acids more stable and protects them from degradation by ribonuclease. 6 With the increasingly extensive application of high-throughput sequencing and microarray technology, noncoding-RNA have been proved to be related to ionizing radiation is increasingly.7-9 It was found that circRNA could be used as microRNA sponge, protein bait and transporter to regulate gene expression. 10 Existing evidence shows that both cyclic ribonucleic acid and micro ribonucleic acid are involved in the pathogenesis and progress of ionizing radiation damage, and play an important role in them.11-13
At present, there are few studies on the characteristics of regulatory network of circRNA-miRNA-mRNA in blood samples of radiation workers. Therefore, this study was designed to investigate the potential circRNA-miRNA-mRNA regulatory network in the peripheral blood of radiation workers. Genome-wide cyclic ribonucleic acid and gene microarray analysis was performed to screen out differentially expressed cyclic ribonucleic acid and genes. Through the ionizing radiation mined in GEO database and the change of micro ribonucleic acid in peripheral blood, the differentially expressed micro ribonucleic acid can be obtained. Prediction and construction of regulatory network of circRNA-miRNA-mRNA. Gene function enrichment analysis of differential genes was conducted to identify their potential function. Our results provide new information on the role of the regulatory network of circRNA-miRNA-mRNA in the health examination of radiation workers and provide a theoretical basis for the interaction mechanism of circRNA, miRNA, and mRNA as biological dose markers and possible disease effects for radiation workers.
Methods
Study Objects
According to the questionnaire scheme in Specification of survey technique for radiation epidemiology, select the appropriate control group and radiation group in the designated hospitals where the industrial radiation workers have occupational health check. 14 With the informed consent form signed, 5 mL of peripheral blood was extracted from the enrolled personnel for subsequent transcriptomic sequencing and RT-qPCR.
Preparation and Sequence of RNA Library
According to the instructions, ribosomal RNA (rRNA) was removed from samples by using Nebner Extranadplenion Kit (New England Bioabs, Inc., Massachusetts, USA). The sequencing library was constructed by using Nebnextremailididential RNAi Podkit (New England BioABS, Inc., Massachusetts, USA. The library was controlled and quantified by the bioanalyzer 2100 system (Agilent Technologies, USA), and 150 bp double-ended sequencing was carried out by Illumina HiSeq instrument.
Differentially Expressed CircRNAs and Differentially Expressed Genes Data Analysis
Sequencing was carried out by an Illumina HiSeq 4000 sequencer, and two-terminal readings were obtained. Q30 was used for quality control, and the cut adapt software (v1.9.3) was used to disconnect and disconnect low quality readings to obtain high quality readings. (1). circRNA: High-quality reads were compared to the reference genome/transcriptome using STAR software (v2.5.1b), and circRNA was detected and identified using DCC software (v0.4.4). The identified circRNA was also annotated with the circBase database(http://circbase.org/) and Circ2Traits. 15 EdgeR software (v3.16.5) was used for data standardization and differential expression. The circRNA passing through fold change>2 and P < .05 was selected as the differentially expressed circRNAs (DECs). (2). mRNA: hisat2 software (v2.0.4) was used to compare high-quality reads to the human reference genome (UCSC HG19). Then, under the guidance of gtf gene annotation file, the FPKM value of mRNA at gene level was obtained as the expression spectrum of mRNA using cuffdiff software (v2.2.1, part of the cufflinks software suite). Fold change >2 and P < .05 were calculated between the 2/group samples to screen for differentially expressed genes (DEGs).
Gene Function Analysis
In order to further understand the function and main functional pathways of mRNA, all the differential functional pathways of mRNA are annotated and analyzed. The mRNA obtained from the above screening was put into David database (https://david.cifcrf.gov/) and the species was selected as “Homo sapiens.” The threshold P < .05 was set for GO enrichment and KEGG pathway annotation analysis, and the enrichment analysis results were visualized.
DEMs in Peripheral Blood After Ionizing Radiation
Microarray data sets of miRNA expression profiles in peripheral blood following ionizing radiation were obtained from the GEO database (https://www.ncbi.nlm.nih.gov/geo/), GSE55954. The miRNA expression profiles of human peripheral blood mononuclear cells in the experimental design at 20 h after exposure to 60 Gy of Cs-137γ-ray were selected. The experiment was carried out independently for 4 times, and the system error was eliminated. The data were analyzed to extract differentially expressed miRNA using the GEO2R program provided by the GEO database. The miRNAs with fold change >2 and P < .05 were selected as differentially expressed miRNAs (DEMs).
Predicting the Target miRNA of DECs and Predicting the Target Genes of DEMs
Using circMir2.0 and mirTargets2.0 software (Nanjing medical university). The software included Starbase (https://starbase.sysu.edu.cn), MIRREDB (http://mirdb.org/cgi-bin), and Target (http://www.targetscan.org) databases to predict circRNA-miRNA and miRNA-mRNA interactions and selected predicted results with an interaction score greater than 80 and a P < .05 for final validation.
Construction of circRNA-miRNA-mRNA Network in Peripheral Blood of Radiation Workers
We extracted the circRNA/miRNA/mRNA competition constraint. The circRNA-miRNA and miRNA-mRNA with potential binding were selected from the predicted differential genes, and the selected DECs, DEMIRs, and DEGs were mapped to construct a circRNA-miRNA-mRNA interaction network, which was visualized using Cytoscape3.8.2 software. 16
RNA Extraction and RT-qPCR
Use HI SCRIPT II first strand cDNA synthesis kit (+GDNA Wiper) kit (Vazyme) to reverse transcribe circrNA and mRNA, and the HI SCRIPT II 1st strand cDNA Synthesis Kit (+GDNA Wiper) kit (Vazyme) were used in reverse transcribed. Use the miRNA first strand cDNA synthesis kit (Bystem-loop) p (Vazyme) to synthesize miRNA reverse transcription kit cDNA. RT-qPCR was used to evaluate the expression of circrNA, miRNA and mRNA, and verified by Chamq universal sybr qpcr master mix kit with PCR instrument (ViiA 7, Life Technologies). GAPDH was used as the internal control for circRNAs and mRNAs, and U6 was used as the internal control for miRNAs. Primers for circRNA and miRNA were designed by CircInteract (https://circinteractome.nia.nih.gov/) and miRNA Design V1 (Vazyme), respectively, and primers for mRNA were found in PrimerBank. The cycle threshold (2-ΔΔct) was compared for calculation. The primers applied are displayed in Table S1.
Statistical Analysis
All statistical analyses were conducted with SPSS 20.0. Independent t test was used to compare continuous variables with normal distribution, Mann–Whitney U test was used to compare continuous variables with skewed distribution, and chi-square test was used to test the ratio. All statistical tests are bidirectional.
Result
Basic Information of People Involved in Gene Sequencing
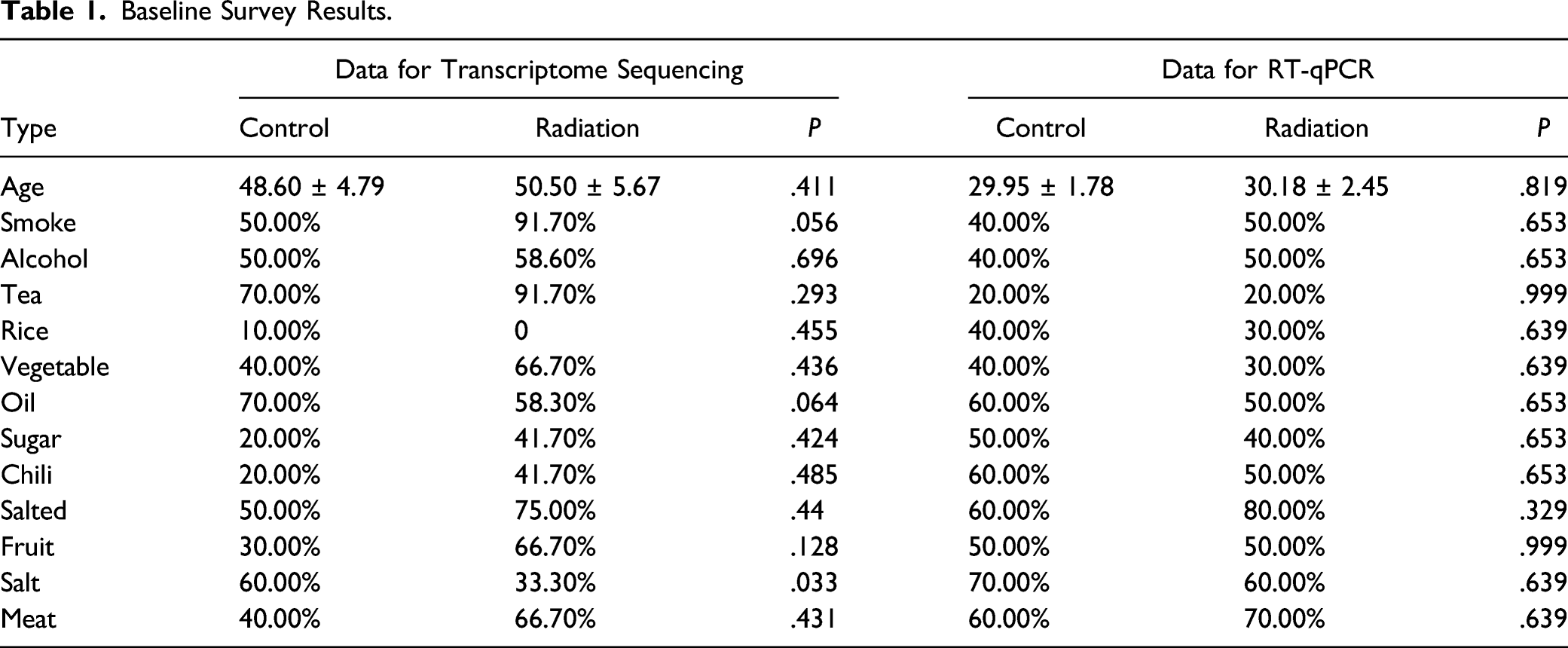
Baseline Survey Results.
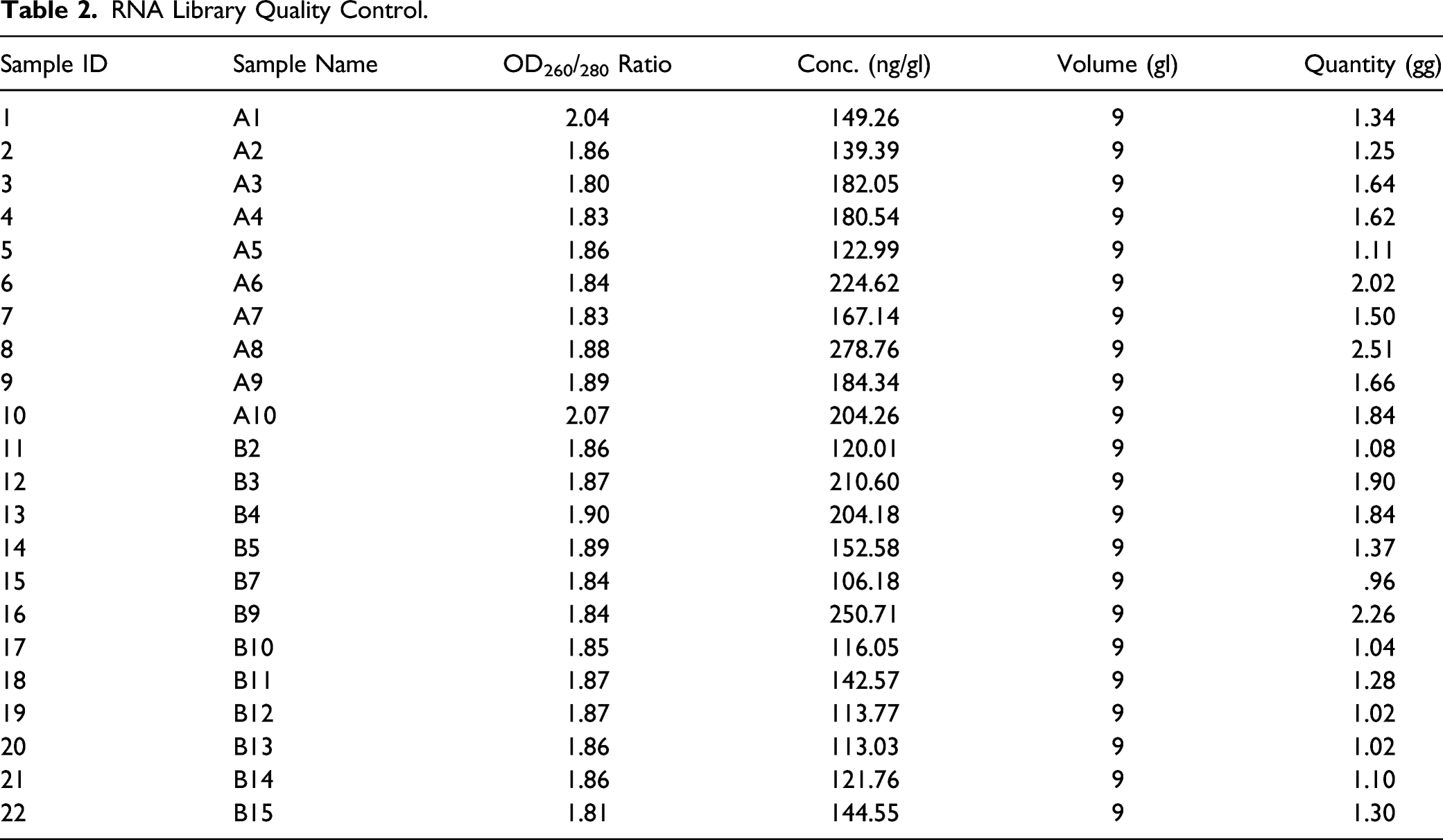
RNA and Library Quality Control
The RNA concentration of each sample was measured using a NanoDrop ND-1000 instrument (Thermo Fisher Scientific, Waltham, MA, USA). The OD 260/OD 280 value was used as the RNA purity index. RNA purity was acceptable when the OD 260/OD 280 value ranged from 1.8 to 2.1; Degenerated agarose gel electrophoresis was used to measure RNA integrity and gDNA contamination, as shown in Figure 1. The quality of the library was measured by Agilent 2100 Bioanalyzer (Table 2). Agarose gel electrophoresis of total RNA. RNA Library Quality Control.
Differential Transcriptome Results
According to the criteria of foldchange>2 and P <.05 between samples, the differential results were screened. Compared with the control group, there were 1149 differentially expressed circRNAs in the radiation group in this study, of which 639 were up-regulated and 510 were down-regulated. A total of 873 differentially expressed genes were identified, of which 68 were up-regulated and 805 were down-regulated. Compared with the control group, in the GSE55954 dataset selected from GEO database, there were 229 differentially expressed miRNA in peripheral blood after irradiation, of which 117 were up-regulated and 112 were down-regulated (Figure 2). Summary of variance expression data. (A): circRNA volcanic map of differential expression between the control and radiology staff blood samples. (B): Volcano map of differentially expressed mRNA between blood samples of radiation workers in the control group and. (C): Volcano diagram showing differential expression of miRNA in peripheral blood after CS-137 γ-ray compared with that in the control group. (D): Summary of up and down regulated genes.
Functional Enrichment Analysis
To understand the biological action and potential function of 873 differential genes, GO analysis was performed, including biological process (BP), cellular component (CC) and molecular function (MF), and KEGG signaling pathway enrichment analysis. GO analysis showed that in terms of BP, these overlapping genes were mainly enriched in the positive regulation of cellular metabolic process, apoptotic process involving in patterning of blood vessels. positive regulation of ERN protein response, cell-substrate junction assembly, positive regulation of cyto-me signaling pathway. For CC, the overlapping genes were mainly enriched in pore complex, MOZ/MORF histone acetyltransferase complex, actomyosin, cyclin-dependent protein kinase holoenzyme complex, and stress fiber. In the case of MF, these overlapping genes were mainly enriched in N-acyl-s phospholipase D activity, phospholipase D activity, G-quadruplex DNA binding,3-chloroallyl aldehyde dehydrogenase activity, and vinculin binding. Enrichment analysis of KEGG signaling pathway revealed that DEGs was mainly enriched in their types of O-glycan biosynthesis, Pertussis, Leishmaniasis, Staphylococcus aureus infection, and Propanoate metabolism (Figure 3). Summary of gene function enrichment. (A): Biological process analysis. (B): Cellular component analysis. (C): Molecular function analysis. (D): KEGG pathway analysis.
Construction of circRNA-miRNA–mRNA Regulatory Network
We obtained the first 20 hub genes using the cytohHubba plug-in of Cytoscape3.8.2 software (Figure 4B), and screened the functional module with the MCODE plug-in to select the first module with the highest score (Figure 4C), and selected a total of 9 genes screened by 2 methods to construct a CIRC RNA–miRNA–mRNA regulatory network (Figure 4D). Finally, we obtained a circRNA-miRNA-mRNA regulatory network consisting of 9 DECs, 3 overlapping miRNAs and 2 target mRNA (Figure 4E). For further details see Table S2. Regulation process of constructing circRNA/miRNA/mRNA network. (A): Analysis of differential gene protein interaction network. (B): top20 hub genes of cytohHubba. (C): Screening first module of MCODE. (D): Total circRNA/miRNA/mRNA network regulation. (E): Regulation of circRNA/miRNA/mRNA network acquired by up-and down-regulation of binding genes. Validation of correlation network regulation in blood samples of radiation workers.
According to the results of RT-qPCR, when compared with the control group, only hsa_circ_0093865 and hsa_circ_0005940 showed no statistical differences in expression levels, and other results were consistent with the results of transcriptomics (Figure 5). RT-qPCR results. (A): the expression level of circRNAs. (B): expression level of miRNAs. (C): mRNA expression level. * means P < .05, and ns means P > .05.
Discussion
Occupational health examination of radiation workers is very important for finding out whether there are defects in their health status in time. For classical examination items such as chromosome micronucleus and chromosome aberration, it can be observed whether the dose is still suitable for the current post. 17 However, it is not clear whether the occupational health of radiation workers will be affected. Epidemiological results show that long-term low-dose ionizing radiation may affect the health of radiation workers, such as cancer, cardiovascular, and cerebrovascular diseases, etc.18,19 Therefore, it is very important to find key molecules and markers. At present, the general peripheral blood examination focuses more on judging the health status of the subjects through the changes of blood cells and blood biochemistry, while the potential genetic changes and changes of non-coding RNA are rarely involved. More and more researchers are involved in the research of non-coding RNA, because these non-coding RNA are involved in the regulation of many diseases, including the research of radioactive diseases and radioactive biodosimeters.
Researchers found that circRNA was associated with many biomarkers of ionizing radiation injury, such as mouse brain and primary neuron injury, mouse bone marrow injury, and radiation-related esophageal injury.20,21,11 In addition, it is also found that circRNA is related to a variety of radiation diseases, such as hsa_circ_0001649 participating in the following regulation of migration and proliferation of cholangiocarcinoma cells after radiotherapy, and hsa_circRNA_ 100367 regulating radiosensitivity of esophageal cancer through Wnt pathway.22,23 The miRNA is an evolutionary conservative small molecule with a length of 18–25 nucleotides. In recent years, there are many studies related to miRNA ionizing radiation, such as miR-29 participating in ionizing radiation-induced fibrosis through the overexpression of inhibitory type I collagen, miR-92b participating in the regulation of susceptibility of liver cancer to ionizing radiation therapy, miR-375-3 p as a marker of acute radiation syndrome, and miR-34 a as a biomarker of ionizing radiation.24-27 These studies show that the research of non-coding ribonucleic acid is very important for ionizing radiation damage and health concerns of radiation workers.
Through literature search, we explored the latest research progress and discovery of NOD2, IRF7, miR-671-5p, and miR-654-5p in circRNA-miRNA-miRNA network that was finally discovered. Nucleotide-binding oligomerisation domain protein 2 (NOD2) participates in studies of ionizing radiation by influencing the regeneration of intestinal epithelial cells damaged by radiation. At the same time, NOD2 alleviates radiation-induced damage through the agonist of ATR-mediated DNA damage response pathway, Morabutanol, and also inhibits oxidative stress and apoptosis by blocking the participation of wow/ROCK-mediated TAK1/NOD2 in NF-κB pathway.28-30 Meanwhile, NOD2 also plays an important role in Parkinson’s disease and intestinal disease.31,32 Interferon regulatory factor 7 (IRF7) is highly expressed in the apoptosis of melanoma cell after ultraviolet irradiation, and it is used as a reactive molecular indicator of radiation-induced injury of human umbilical vein endothelial cells.33,34 Of course, IRF7 has been proved to play a role in virus-induced cellular gene transcription activation, and its encoded protein plays an important role in the innate immune response against deoxyribonucleic acid and ribonucleic acid virus. 35 The miR-671-5p can be used as a biomarker to detect the therapeutic effect of radiotherapy on colorectal cancer, 36 furthermore, the invention can also be used as a new biomarker for early breast cancer detection and a target for breast cancer treatment; It also inhibits the progression of glioblastoma in circ_0001946/miR-671-5p/CDR1 network regulation.37,38 miR-654-5p can target and regulate EPSTI1 to slow the progression of breast cancer; in addition, it can inhibit the MYC, WNT, and AKT pathways from affecting the development of ovarian cancer.39,40 In our study, all circRNAs and miR-3616-3p were both reported for the first time, and we hope to find out their functions through further experiments at the later stage.
In conclusion, we have constructed a circRNA-miRNA-mRNA network by comparing the expression of circRNAs and mRNAs in peripheral blood of radiation workers and healthy control, and the ionizing radiation-related miRNAs obtained from the GEO database. Subsequently, I constructed a circRNA-miRNA-miRNA network consisting of 9 circRNAs (And the expression levels of the 7 circRNAs were consistent with transcriptomics), 4 miRNAs and 2 hub genes using a variety of different bioinformatics methods. This research can provide some help for the development of biological dosimeters for future dose estimation of radiation workers and the possible mechanism of radiation-related diseases. Of course, this needs further experimental verification in the later period.
Supplemental Material
Supplemental Material - Analysis of circRNA-miRNA-mRNA Regulatory Network in Peripheral Blood of Radiation Workers
Supplemental Material for Analysis of circRNA-miRNA-mRNA Regulatory Network in Peripheral Blood of Radiation Workers by Jin Gao, Tinxi Lan, Xumin Zong, Gensheng Shi, Shuqing He, Na Chen, Fengmei Cui, and Yu Tu in Journal of Dose-Response
Supplemental Material
Supplemental Material - Analysis of circRNA-miRNA-mRNA Regulatory Network in Peripheral Blood of Radiation Workers
Supplemental Material for Analysis of circRNA-miRNA-mRNA Regulatory Network in Peripheral Blood of Radiation Workers by Jin Gao, Tinxi Lan, Xumin Zong, Gensheng Shi, Shuqing He, Na Chen, Fengmei Cui, and Yu Tu in Journal of Dose-Response
Footnotes
Acknowledgments
We thank CNNC 401 Hospital for the great help. Thanks to my dear Duobao for her support.
Authors’ Note
Conceived and designed the experiment: Yu Tu. Performed the experiments: Jin Gao, Tinxi Lan, Xumin Zong, Gensheng Shi, Shuqing He, Na chen, Fengmei Cui. Analysed the data: Jin Gao Tinxi Lan and Xumin Zong. Wrote the paper: Jin Gao.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Natural Science Research Projects of Colleges and Universities in Jiangsu Province (grant numbers 20KJB310007), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the Collaborative Innovation Center of Radiological Medicine of Jiangsu Higher Education Institutions, China and the Nuclear Energy Development Project, China (No. 2016-1295).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.