
Abstract
Background:
Mounting evidence suggests that alternative splicing is one of the ways for cells to adapt to environmental stress insults. The aim of this study was firstly to examine the effect of silica on the alternative splicing of lung fibrosis–associated genes.
Methods:
Microarray analysis was used to construct the alternative splicing profile. Functional experiments were conducted using Cell Counting Kit-8, cell cycle, apoptosis, and epithelial–mesenchymal transition (EMT) analyses. Alternative splicing variants were verified by quantitative real-time polymerase chain reaction (qRT-PCR) polymerase chain reaction method.
Results:
A total of 1850 genes that have alternative splices in response to silica insult were identified. PCDHB11, MALAT1, MT2A, RP11-126D17.1, and RP11-415I12.2 are the top 5 upregulated genes with occurrence of alternative splice, whereas NDE1, RNPEPL1, TREML2, CSF2RB, and PRKCSH are the top 5 downregulated genes with occurrence of alternative splice. Bioinformatic analysis showed these genes with the occurrence of alternative splice mainly are associated with EMT pathway, N-Glycan biosynthesis, and leukocyte transendothelial migration. Further study indicated that PRKCSH-2 knockdown promotes A549 cell proliferation potential by partially promoting EMT signals.
Conclusions:
Significant changes in alternative splicing of silicosis-associated genes occur in patients with silicosis in silica conditions. Our study provides basic founding for further investigation into the detail molecular mechanisms underlying silica-induced silicosis.
Introduction
Environmental or occupational exposure to mineral dusts, mainly silica, is associated with an increased incidence of lung inflammation, fibrosis, and/or cancer. Silica, also known as silicon dioxide, is the main component of more than 95% of earth rocks.
1
Workers exposure to silica dust with diameter less than 10 µM via respiratory system inhalation and deposition mainly result in silicosis, a fibrotic respiratory disease with major pathological presentation of progressive lung fibrosis.
2,3
Indeed, silicosis is not a newly identified disease.
4
During the period of 370 to 430
PRKCSH (Protein kinase C substrate 80K-H), firstly isolated by Sakai et al in 1989, 13 located on chromatin 19p13.2 consists of multiple domains including translocation sequence, an N-terminal GIIα-binding domain, a glutamic acid, and proline-rich segment and a C-terminal mannose 6-phosphate receptor homology domain. 14 The Project of Analysis Human Tissue revealed that higher expression level exists in normal lung, thyroid, placenta, and ovary tissues. 15 We searched the website of https://www.uniprot.org/uniprot/P14314 and found that PRKCSH is mainly involved in the cellular protein metabolic process, protein folding, posttranslational protein modification, and so on. Numerous studies have reported the multiple functions of PRKCSH. Shin et al indicated that PRKCSH contributes to tumorigenesis by selective boosting of IRE1 signaling pathway. 16 Mutations in PRKCSH including heterozygous duplications, small deletions, missense, and splice site mutations have been identified 17 and associated with certain diseases such as autosomal dominant polycystic liver disease. 17 Furthermore, the expression alteration was found to be associated with the progression of breast cancer lymph node metastasis. 18 However, the function of PRKCSH in the silica-induced lung fibrosis are not fully understood. Here, in this study, we demonstrate a new role of PRKCSH alternative splicing (AS) of pre-messenger RNA (mRNA) products in the regulation of progression of epithelial–mesenchymal transition (EMT) in the lung epithelial cells.
Alternative splicing is a ubiquitous regulatory mechanism of gene expression that allows generation of more than one unique mRNA species from a single gene. 19,20 The occurrence and molecular mechanism of AS are through generating different mRNAs in untranslated regions or in the coding sequence through exon skipping, selection between mutually exclusive exons, the usage of alternative splice sites, and intron retention. 20 As a result of AS, mRNA stability, location, and translation can be influenced. 21 Moreover, some AS events generate different protein isoforms with different dramatically functions or localization. 22 Analysis of 15 diverse human tissue indicated 92% to 94% of human genes undergo AS, and 86% with a minor isoform frequency of 15% or more. 23 The research of AS has progressed gradually currently from identifying AS events to their impact on protein expression and how they are molecularly coordinated. Some studies were also performed to investigate the AS events in the progression of lung fibrosis. Chen et al explored AS of fibroblast growth factor receptor 2 (FGFR2) in bleomycin (BLM)-induced lung fibrotic and transgenic mouse models. The results showed BLM inhibited the expression of epithelial splicing regulatory protein 1, resulting in enhanced AS of FGFR2 to the mesenchymal isoform IIIc. 24 Nance et al reported that 440 unique genes had significant differential splicing events in at least one exonic region through transcriptome analysis from 8 idiopathic pulmonary fibrosis lung samples. 25 Zhang et al identified a mineral dust-induced gene named mdig and detected alternative spliced transcripts of this gene in A549 lung cancer cells. 26 Therefore, increasing evidence certainly demonstrates that AS contributes to the regulation of cell function through proteome complexity. However, extensive further work is needed to identify the functional consequences of AS events in the condition of certain stress stimulation. Here, based on our laboratory studies on the transcriptomics of silica-induced lung fibrosis previously, 11 we noted an increased AS events in the silica-induced lung fibrosis. Bioinformatic analysis and characterization of these AS events uncovered some genes involved in significant AS events. After verification and a series of phenotype experiments, two PRKCSH AS transcripts (PRKCSH-1 and PRKCSH-2) have different functions, in particular, in the regulation of EMT progression. Thus, in this study, we demonstrate a molecular understanding of AS occurring during the silica-induced silicosis development, suggesting PRKCSH alternative splice transcripts could be the prevention or therapy targets in silica-induced lung fibrosis.
Methods and Materials
Human Peripheral Blood Samples Collection and Total RNA Isolation
Ten participants were divided into 2 groups, 5 participants who were exposed to silica with identified phase I silicosis were the case group and 5 healthy participants who worked in the same industry but were not exposed to silica were the control group. Details of the baseline information including the inclusion criteria, environmental silica concentration exposure, mean weight, and the blood sample storage condition are presented in our previous study. 11 The study was performed in accordance with all relevant tenets of the Declaration of Helsinki and was approved by the Ethics Committee of Xiangya School of Public Health (Approval no. XYGW-2018-11). All participants provided written informed consent. Total RNA isolation was performed according to the instruction of mirVanaRNA Isolation Kit. 11
Microarray Analysis
After total RNA were isolated from the 2 groups, the mRNA expression was assayed using the Affymetrix GeneChip Human Transcriptome Array 2.0 platform at Beijing Compass Biotechnology Co, Ltd. The Transcriptome Analysis Console 3.0 software was used for AS analysis. Filter criteria were used based on the Affymetrix recommendation including adjusted false discovery rate (FDR) P value and splicing index. False discovery rate P value means FDR-corrected P value. We calculated FDR P value using Benjamini-Hochberg method. 27 The splicing index was calculated as:

FDR P value <.05 and splicing index >2 were considered to be significance in AS events.
Quantitative Real-Time Polymerase Chain Reaction Validation
Quantitative real-time polymerase chain reaction (qRT-PCR) method was used to further verify the AS genes in extended other samples. In brief, this method is conducted using PEXBIO RNA kit according to the manufacturer’s protocol (CFX96TM Real-Time System; Bio-Rad). Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) served as the internal control and fold-change calculation were made using the 2–Δ ΔCT method, which is calculated as:

Ct means cycle at which the threshold is reached. Primer sequences for qPCR are listed in Table 1.
Primers Used for Detection of Selected 10 Genes with Various Alternative Splicing Transcripts.

Cell Culture, Treatment, Small Interfering RNA, and Transfection
HBE cells were purchased from ATCC (American type culture collection, www.atcc.org/) and A549 cells were purchased from the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences. 28 A549 and HBE cells were cultured in RPMI-1640 medium (Thermo Scientific) supplemented with 10% fatal bovine serun (FBS). After the cells were more than 85% confluent, they were cultured in serum-free medium for approximately 6 hours, and transforming growth factor beta 1 (TGF-β1; Merck KGaA) at a final concentration of 10 ng/mL was then added to treat the cells. Total RNA and protein were collected at different time points. The levels of PRKCSH and EMT-related biomarkers including E-cadherin, N-cadherin, vimentin, and α-smooth muscle actin (α-SMA) were analyzed by qRT-PCR and Western blotting.
To knockdown the PRKCSH expression, small interfering RNA (siRNA) duplexes were designed and synthesized by (Shanghai GenePharma Co, Ltd). The 2 PRKCSH different AS transcripts’ sequences are listed as follows: PRKCSH-1: 5*…….ATGGAGAACCAAGGGACACG…….3* and 5*…….CTAAAAATTAAATCCAAAGC…….3*, PRKCSH-2: 5*…….ATGGTTTTTAGTATCCAAGA…….3* and 5*…….CTAAAAATTAAATCCAAAGC…….3*. Four nanometre (nM) siRNA duplexes were transfected into cells using Lipofectamine RNAiMAX (Invitrogen).
Cell Proliferation
Cell proliferation ability was estimated through Cell Counting Kit-8 (CCK-8) and wound healing experiments. Cell Counting Kit-8 was performed according to the manufacturer’s instruction. Wound healing assay was conducted as follows: HBE cells were seeded into 6-well plates (4 × 105 cells/well), and 2% FBS-supplemented medium was added to avoid cell proliferation before incubation at 37 °C for 24 hours. si-NC (NR) and si-PRKCSH were transfected into cells. Then, freshly changed 2% FBS-supplemented medium was added after the medium was removed, and wounds were created with a sterile 200-µL pipette tip in each well. Wound healing was monitored and photographed at indicted groups.
Western Blot Analysis
The antibodies of EMT-related biomarkers including E-cadherin, N-cadherin, Vimentin, and α-SMA were purchased from Cell Signaling Technology. The antibody of PRKCSH was purchased from Santa Cruz Biotechnology. Briefly, the samples were treated with lysis buffer, subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis, and transferred to polyvinylidene fluoride membranes. After the membranes were blocked with 5% nonfat milk in Tris-buffered saline containing Tween-20 for 2 hours, the membranes were incubated with primary and secondary antibodies overnight at 4 °C. An ImageQuant LAS500 system (Molecular Dynamics) was used to visualize the bands. Details of the Western blot analysis can be found in our previous publications. 28 -31
Apoptosis Detection
In this study, apoptosis was assessed using a fluorescein isothiocyanate (FITC)-Annexin V Apoptosis Detection Kit (BD Pharmingen) following the manufacturer’s instructions. The cells were divided into different groups based on the design requirement. After the cells were harvested, the cells were centrifuged, resuspended in phosphate-buffered saline (PBS), and transferred to clean Eppendorf tubes, and 100 µL of 1× Annexin V-binding solution was then added to the cells to form a suspension of 1 × 10 cells/mL. Subsequently, 5 µL of FITC-conjugated Annexin V and propidium iodide (PI) solution (40 μg/mL PI and 0.1% Triton X-100 in PBS buffer) was added. The cells were then incubated for 15 minutes at room temperature in the dark, and 400 µL of 1× binding buffer was then added. The cells were then analyzed by flow cytometry. 32
Cell Cycle Analysis
The cells were seeded into 35-mm culture dishes at a density of 70% to 80% per dish. The cells were divided into different groups based on the design requirement. After the cells were harvested, the cells were treated with RNase A and incubated at 37 °C for 30 minutes. The cells were stained with PI solution, and the cell cycle distribution was analyzed by flow cytometry.
Statistical Analysis
All data calculations were performed using SPSS version 18.0 software (SPSS, Inc), and figures were plotted applying GraphPad Prism version 5.0 software (GraphPad). Continuous variables were shown as average ± standard deviation, and their differences between the 2 groups were analyzed through Student t test. 33
Results
Silica Insult Increases Levels of Alternative Splice Events in Patients With Silicosis
We previously demonstrated that silica exposure causes significantly transcriptomics alteration in patients with silicosis compared to the normal population. 11 While characterizing the transcriptome after silica insult, we detected the AS events were increased in the group of patients with silicosis. A total of 2206 AS events were identified, of which 887 were intron retention, 559 were cassette exon, 403 were alternative 3′ acceptor site, and 357 were alternative 5′ donor site. A total of 1850 genes have alternative splices in response to silica insult, based on the filter criteria, exon splicing index >2, and exon FDR P value <.05. As shown in Figure 1A and listed in Table 1, compared to the control group, 1604 multiple complex (gene contains more than one locus type above; 86.7%), 190 noncoding (10.27%), 40 coding (2.16%), 13 unassigned (0.7%), and 3 (0.16%) pseudogenes were identified (Table 2). Details of genes with AS events are listed at Table 3. As the list with top 20 changed genes, PCDHB11, MALAT1, MT2A, RP11-126D17.1, and RP11-415I12.2 are the most upregulated gene with fold-change beyond 3 times and exon splicing index >2, suggesting these top 20 genes have the significant chance to subject to the AS. Of the list with top 20 changed genes, NDE1, RNPEPL1, TREML2, CSF2RB, PRKCSH are the most downregulated genes with fold-change beyond 3 times and exon splicing index >2, suggesting these top 20 genes have the significant chance to subject to the AS.

Bioinformatic analysis of alternative splice transcripts. A, Based on the filter criteria (P < .05, splice index >2), the resource of alternative splicing transcripts distribution percentage. B, Heatmap of alternative splice transcripts; patients with silicosis is in red, whereas normal control individuals are in blue. C. A volcano plot of alternative splice transcripts; the upregulated expression of transcripts are in red, whereas the downregulated expression of transcripts are in green.
Potential Alternative Splicing Genes.

Abbreviation: FDR, false discovery rate.a Filter criteria: exon splicing index: < −2 or > 2, exon FDR P < .05.
Top 20 Differential Expressed Genes Based on P < .05 and Splicing Index >2 in Patients With Silicosis Compared With Normal Group.a

a P < .05 represents significantly changed. Splicing index = (exon 1 condition 1 intensity/gene 1 condition 1 intensity)/(exon 2 condition 2 intensity/gene 2 condition 2 intensity); FDR: false discovery rate corrected P value.
Heatmap illustrator using unsupervised cluster analysis of alternative splice genes of the 10 samples, in terms of differentially expressed genes, yielded the results illustrated in Figure 1B. Highly expressed alternative splice genes are shown in red, whereas alternative splice genes expressed in low level are shown in blue. A volcano plot map of these genes with possible alternative splice is also shown in Figure 1C. Highly expressed alternative splice genes are shown in red, whereas alternative splice genes expressed in low level are shown in green. Heatmap and volcano plot suggested that the upregulated and downregulated genes have the chance to subject to alternative splicing in response to silica insult.
Bioinformatic Analysis Reveals the Involvement of Top 10 Genes in the Biological Function
To explore the possible function of the genes with alternative splicing in response to silica exposure, we performed bioinformatic analysis of GO (Gene Ontology resource) and KEGG (Kyoto Encyclopedia of Genes and Genomes) according to our previous reports. 34 Gene Ontology analysis consists of 3 kinds: biological process, cell component, and molecular function, and the analysis was conducted on the website of www.geneontology.org/. The result of GO analysis (Figure 2A) shows that during the biological process, top 10 genes with alternative splice transcripts were involved in positive regulation of transcription, regulation of phosphoprotein, and apical junction assemble, whereas in the cell component, they mainly involved in the nuclear membrane, intracellular ribonucleoprotein complex, and nucleus, whereas in molecular function, RNA binding, transcription coactivator activity, and GTPase regulator activity are the mainly predicted function of these top 10 genes. Kyoto Encyclopedia of Genes and Genomes analysis is a database resource for understanding high-level functions and utilities of the biological system, such as the cell, the organism, and the ecosystem, from molecular-level information, especially large-scale molecular data sets generated by genome sequencing and other high-throughput experimental technologies (https://www.kegg.jp/). As shown in Figure 2B, EMT pathway, N-Glycan biosynthesis, and leukocyte transendothelial migration are the mainly pathways of these top 10 genes. These data indicated that these top 10 genes with alternative splice transcripts in response to silica may involve multiple cell signaling pathways and molecular mechanisms.

Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of alternative splice transcripts. A, The GO analysis of top 10 significantly altered alternative splice transcripts, consisting of biological process, cell component, and molecular function. B, The KEGG analysis of top 10 significantly altered alternative splice transcripts.
Verification of Expression Changes in Genes With Alternative Splice
To determine whether the microarray analysis of alternative splice can be verified, 7 genes were selected randomly to perform qRT-PCR in 20 cases and 20 controls: ANKRD52, MFSD6, NDE, GPR34, PRKCSH, MAPK1IP1L, and NUP210. The expression levels of cases and controls differed significantly (Figure 3A-G), consistent with the microarray data, indicating the microarray analysis results can be used for further function and molecular study. Furthermore, we also investigated the expression level of different AS transcripts in response to silica. For instance, ANKRD52 alternative splice transcript-2 was significantly upregulated in the case group, whereas alternative splice transcript-1 without significant alteration in the case group compared to the control group. Two alternative splice transcripts of MESD6, NUP210, and GPR34; 3 alternative splice transcripts of NDE; and 4 alternative splice transcripts of PRKCSH were all significantly altered in the case group compared to the control group. Here, we focused on the 4 alternative splice transcripts of PRKCSH to the further study as this gene has a high expression in normal human lung tissue. 15 Therefore, A549 cells were used to determine the expression levels of 4 AS transcripts of PRKCSH. As shown in Figure 3H, PRKCSH-1 was significantly regulated in response to silica exposure, whereas PRKCSH-2 was significantly downregulated in response to silica exposure. These data indicated that PRKCSH-1 and PRKCSH-2 may have different regulation function in the lung cell response to silica. Considering that GO and KEGG analysis showed that top 10 genes may be involved in the EMT pathway, N-Glycan biosynthesis, leukocyte transendothelial migration, and so on, we then conducted a series phonotype experiments in HBE and A549 cells.

Verification of splice variants by qRT-PCR in blood samples from patients with silicosis and normal control population. A, Verification of ANKRD52 splice variants by qRT-PCR in blood samples from patients with silicosis and normal control population. B, Verification of MFSD6 splice variants by qRT-PCR in blood samples from patients with silicosis and normal control population. C, Verification of NDE splice variants by qRT-PCR in blood samples from patients with silicosis and normal control population. D, Verification of GPR34 splice variants by qRT-PCR in blood samples from patients with silicosis and normal control population. E, Verification of PRKCSH splice variants by qRT-PCR in blood samples from patients with silicosis and normal control population. F, Verification of MAPK1IP1L splice variants by qRT-PCR in blood samples from patients with silicosis and normal control population. G, Verification of NUP210 splice variants by qRT-PCR in blood samples from patients with silicosis and normal control population. H, Determination of PRKCSH splice variants by qRT-PCR in A 459 cells with or without treatment with silica. I, Summary information of 2 PRKCSH splice variants, PRKCSH-1(ENST0000058750.5) and PRKCSH-2 (ENST00000593053.1), available on website of http://libdb.csu.edu.cn/rwt/PUBMED/http/MF3XTZJPMWYHG3LNMJXC655TMH/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000130175;r=19:11435635-11450968;t=ENST00000592741.
We searched the website of Ensembl (http://asia.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000130175;r=19:11435288-11450968) using the keyword PRKCSH and found there exists 20 transcripts (splice variants) of this gene, of which 9 are protein coding, 1 is nonsense-mediated decay, 3 are processed transcript, and 7 are retained intron. In this study, we identified 4 splice transcripts, of which 2 transcripts have significant alteration of expression in A549 cells in response to silica exposure. One transcript, named as ENST00000587509.5 (PRKCSH-206) with coding protein of 140aa, is consistent with that reported previously on the Ensembl website. Another transcript, named as ENST00000593053.1, is a processed transcript with transcript length of 588 bp.
PRKCSH-2 Deficiency Promotes Cell Proliferation
To explore the function of PRKCSH-1 and PRKCSH-2 in lung cancer cell progression, A549 cells treated with TGF-β (Figure 4A) were taken as the case group. A549 cells were transfected by siPRKCSH-1 and si-PRKCSH-2, respectively, to inhibit their expression. Cell Counting Kit-8 assay was used to detect the cell proliferation; the result demonstrated that transfection with siPRKCSH-2 significantly promoted the cell proliferation (Figure 4B). Wound healing assay further confirms the role of PRKCSH-2 in the promotion of cell proliferation (Figure 4C and D). Furthermore, we continue to determine the function of PRKCSH-1 and PRKCSH-2 in regulation of lung cell cycle using flow cytometry (Figure 4E and F). There was no significant difference in cell cycle in siPRKCSH-1 and siPRKCSH-2 cells compared with the control group; these data suggested that PRKCSH-1 and PRKCSH-2-mediated cell proliferation may be due to other pathways.
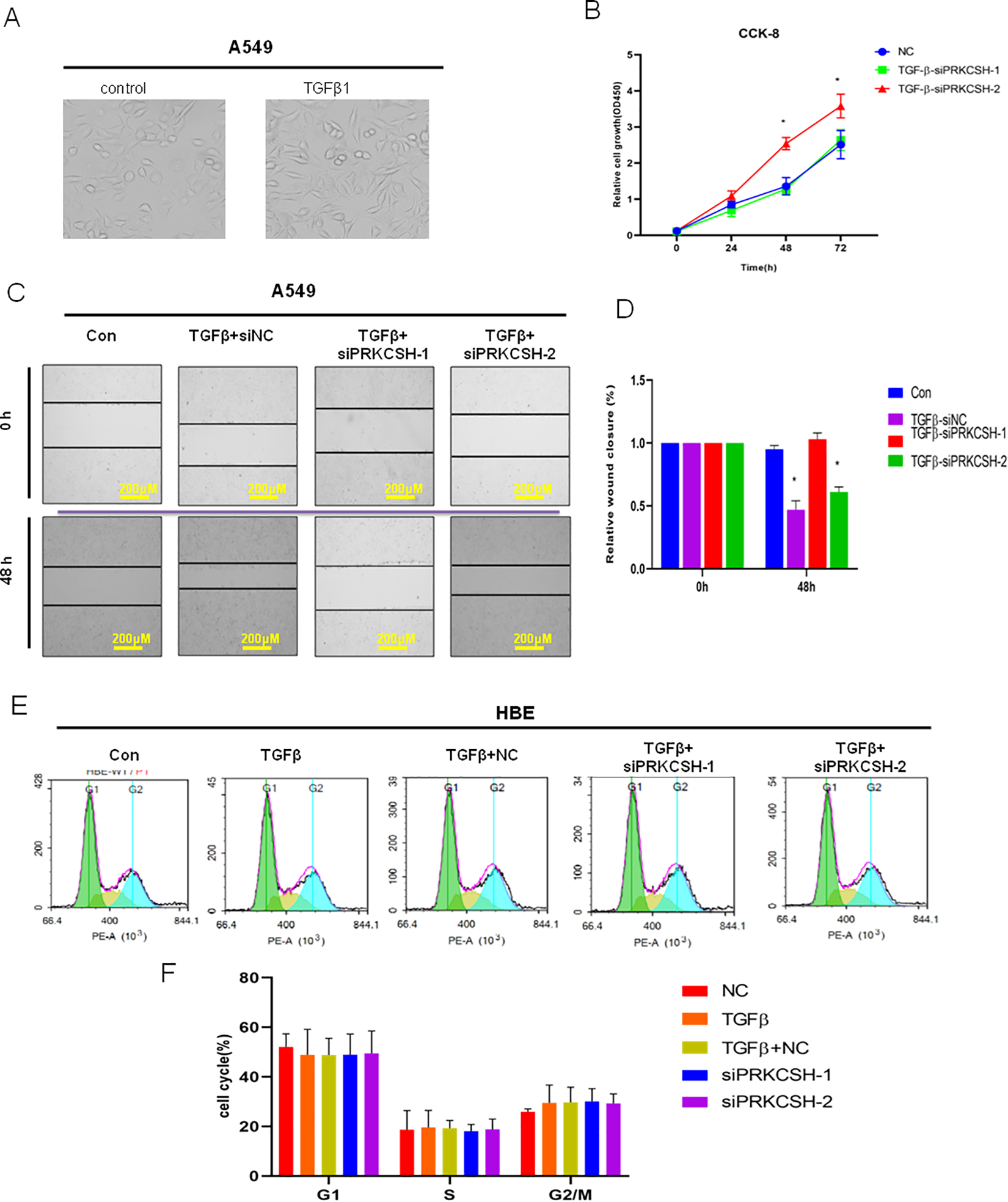
PRKCSH-2 deficiency promotes lung cancer cell proliferation. A, Representative image of lung A549 cells with or without TGFβ treatment (magnification, ×40). B, Cell Counting Kit-8 (CCK-8) assay was used to determine the effect of silencing the PRKCSH splice variants on the cell proliferation. C, Wound healing assay showed that silence of PRKCSH-2 significantly repressed cell migration ability of A549 cells (magnification, ×10). D, Quantitative determination of wounding healing results. The results represent the average of 3 independent experiments (mean ± standard error of the mean); *P < .05. E, Flow cytometric analysis of cell cycle in 5 groups, control group, TGFβ treatment, TGFβ treatment + negative control (NC), TGFβ treatment + siPRKCSH-1, and TGFβ treatment + siPRKCSH-2. F, Quantitative determination of wounding healing results. The results represent the average of 3 independent experiments (mean ± standard error of the mean); *P < .05.
PRKCSH-2 Knockdown Promotes A549 Cell Proliferation Potential by Partially Promoting EMT Signals
Subsequently, whether PRKCSH-1 and PRKCSH-2 affect the cancer cells apoptosis was investigated. There was no significant difference in apoptosis in the PRKCSH-1 and PRKCSH-2 knockdown A549 cells compared with the control group (Figure 5A and B). To test the effect of PRKCSH-1 and PRKCSH-2 knockdown on cell proliferation associated with EMT, Western blotting experiment was used to determine the expression of EMT-related biomarkers. As shown in Figure 5C and D, PRKCSH-1 knockdown increased epithelial biomarker E-cadherin expression and decreased expression levels of mesenchymal biomarkers Vimentin, E-cadherin, and α-SMA in A549 cells compared with the group treated with TGFβ, whereas PRKCSH-2 knockdown decreased epithelial biomarker E-cadherin expression and increased expression levels of mesenchymal biomarkers Vimentin, E-cadherin, and α-SMA in A549 cells compared with the group treated with TGFβ. These data indicated different PRKCSH alternative splice variants of lung cancer cell proliferation via differentially modulating the EMT signaling pathway.

PRKCSH-2 deficiency promotes TGFβ-induced EMT progression. A, Flow cytometric analysis of apoptosis in 5 groups, control group, TGFβ treatment, TGFβ treatment + negative control (NC), TGFβ treatment + siPRKCSH-1, and TGFβ treatment + siPRKCSH-2. B, Quantitative determination of wounding healing results. The results represent the average of 3 independent experiments (mean ± standard error of the mean); *P < .05. C, Western blotting was used to determine the effects of silence PRKCSH-1 and PRKCSH-2 on the TGFβ-induced EMT progression. D, Quantitative determination of wounding healing results. The results represent the average of 3 independent experiments (mean ± standard error of the mean); * P < .05 EMT indicates epithelial–mesenchymal transition; TGFβ, transforming growth factor beta 1.
Discussion
In this study, we demonstrate that silica induce a number of alternative splice variants expression in patients with silicosis compared to the normal population. Specifically, we identified the expression levels of PRKCSH-1 and PRKCSH-2, 2 PRKCSH transcript variants, were altered in response to silica insults. Furthermore, loss of PRKCSH-2 promotes lung cancer cell proliferation via regulating EMT pathway with decreased epithelial biomarker E-cadherin expression and increased expression levels of mesenchymal biomarkers Vimentin, E-cadherin, α-SMA in A549 cells. Our data indicate that PRKCSH gene is subject to AS, as supported by the findings presented above. More specifically, 2 splice variants of PRKCSH showed a different expression pattern in response to silica and perform different regulation functions in the expression of TGFβ-induced EMT markers. Our results are consistent with regulated AS a mechanism for altered PRKCSH expression in response to environmental stress. 35,36 Moreover, this study highlights a previously unrecognized mechanism for silica-induced PRKCSH transcript variants and points toward its potential involvement in silicosis initiated by silica insult.
Promoted by the rapid development of high-throughput genomic profiling technologies, 37 the potential of alternative splice expression alteration under various environments has been increased recently. 38,39 Athman et al took a computational analysis of AS across mammalian tissues and revealed rhythmic AS events are widespread across mammalian tissues and might contribute to a temporal diversification of the proteome. 39 Chen et al identified a DNA damage-induced AS pathway which can regulate p53 and cellular senescence markers using microarray analysis. 40 Using the same technique, Macaeva et al identified radiation-induced alternative transcription and splicing events, as well as these transcript variants’ role in the applicability to practical biodosimetry. 41 In a recent report, Collier et al indicated that when cells in response to sublethal ultraviolet B radiation, genome-wide analysis reveals preferential translation of a series of genes undergoing AS events following radiation, of which the CDKN1A splice variants regulated the fate of radiation-insulted human keratinocytes. 42 Other environmental stresses, such as hypoxia, have been also found to induce the occurrence of alternative events. As stated in the study reported by Bowler et al, hypoxia leads to significant changes in AS in prostate cancer cells and the CLK splice factor kinase could be targeted in cancers in which hypoxia contributes to cancer therapy resistance. 43 Following these reports, we used the microarray method to screen the alternative splice variants in the patients with silica-induced silicosis compared with the normal population and identified a number of alternative splice variants. Our study further indicates that silica, a common environmental and occupational factor, can induce alternative splice variants, and these variants may be associated with silica-induced EMT progression.
As we illustrated in the Results section, till now, 20 transcripts (alternative splice) of PRKCSH have been reported previously, some of which are protein coding and some of which are processed transcript (http://asia.ensembl.org/). Here, in this study, 4 transcripts of PRKCSH have been found with altered expression in response to silica. We further found 2 (PRKCSH-1, ENST00000587509.5 and PRKCSH-2, ENST00000593053.1) of the 4 transcripts have different function in regulation of cancer cell proliferation through affecting the TGFβ-mediated EMT markers. Drenth et al indicated a splice-acceptor site mutation (1138-2A→G) in PRKCSH in 3 families and a splice-donor site mutation (292+1G-->C) in PRKCSH as the probable cause of polycystic liver disease. 44 Waanders et al identified a total of 26 novel PRKCSH mutations using a combination of splice site recognition and evolutionary conservation and found many mutations were predicted to affect splice. 45 In addition, PRKCSH contributes to tumorigenesis. 16 In fact, due to the high expression distributed in lung and other tissues, PRKCSH splice transcripts may perform regulation roles more widely in biological process. Here, we identified the novel roles of 2 PRKCSH transcripts (alternative splice) promote the proliferation of lung cancer cells via regulation of EMT markers. The results in this study also showed that the cell cycle and apoptosis were not affected by the PRKCSH alternative splice; this may be due to the other factors or molecular mechanisms which need further investigation.
However, extensive further work is needed to identify the functional consequences for most of the identified splicing events. In addition, not all detected AS events might necessarily result in the production of functional proteins.
There are still a lot of issues needed to be elucidated in the future. (1) As the AS is a critical component of the regulation of gene expression pathways, how the detailed mechanisms and regulation of alternative pre-mRNA splicing should be elucidated. (2) Since many splice factors are involved in multiple hallmarks of diseases, 46 investigation on these factors might provide a new layer of disease treatment or a potential method to combat disease progression. (3) Why different transcripts perform different functions? Who is the master to control this performance? This issue should be further studied. (4) Investigation on the network of splice variants, RNA binding proteins, and relative cis elements would further shed light on the emergence, mechanisms, and functions of splicing networks. Meanwhile, it also needs to be considered that nucleic acid amplification methods can generate some artifacts, including overestimation of splicing events.
We recognize that there are few limitations in our study. First, it is that our results are based on microarray prediction and bioinformatic analysis, where the enrolled participants were only 5 samples in each group, and the number of the participants should be increased in the further study. Second, the function of PRKCSH transcripts was only investigated in the knockdown condition rather than overexpression of the PRKCSH transcripts. To address this, we will construct the overexpression plasmid to further verify the function of PRKCSH transcripts.
Taken together, we have shown that alternative splice events occur in patients with silicosis. These alternative splice variants generated by silica stimulation were predicted to involve into silica-induced biological process, molecular functions, and signaling pathways. Implementation of TGFβ-induced EMT experiments showed PRKCSH-1 transcript promotes the EMT progression. These data contribute to improve our understanding of silica-related changes in expression of PRKCSH and its splice transcripts involved in the regulation of cancer cell proliferation through changing expression of EMT-related markers.
Footnotes
Authors’ Note
P.-K.Z. contributed to the study concept and critical design of the study. R.X.H., L.H., and X.D.L. conducted the cell experiments. R.X.H. analyzed and interpreted the data. R.X.H. wrote the initial manuscript. P.K.Z. critically reviewed and revised the final manuscript. All the authors read and approved the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from the National Natural Science Foundation of China (Grant Nos. U1803124, 31530085, 31870847, and 81842033).