
Abstract
Preplacement of compensatory tissue repair (CTR) by exposure to a nonlethal dose of a toxicant protects animals against a lethal dose of another toxicant. Although CTR is known to heteroprotect, the underlying molecular mechanisms are not completely known. Here, we investigated the mechanisms of heteroprotection using thioacetamide (TA): acetaminophen (APAP) heteroprotection model. Male Swiss Webster mice received a low dose of TA or distilled water (DW) vehicle 24 hours prior to a lethal dose of APAP. Liver injury, tissue repair, and promitogenic signaling were studied over a time course of 24 hours after APAP overdose to the TA- and DW-primed mice (TA + APAP and DW + APAP, respectively). Thioacetamide pretreatment afforded 100% protection against APAP overdose compared to 100% lethality in the DW + APAP-treated mice. Although hepatic Cyp2e1 was similar at the time of APAP administration, immediate activation of hepatic c-Jun N-terminal kinases (JNK) was observed in the TA + APAP-treated mice compared to its delayed activation in the DW + APAP group. In contrast to the DW + APAP group, the TA + APAP-treated mice exhibited extensive CTR, which was secondary to the timely activation of Wnt/β-catenin pathway. Our data indicate that rapid activation and appropriate termination of Wnt/β-catenin signaling and modulation of JNK activity underlie TA + APAP heteroprotection.
Introduction
It is widely known that low to moderate doses of hepatotoxicants such as acetaminophen (APAP), carbon tetrachloride, and thioacetamide (TA) cause minimal liver necrosis in mice and rats. 1 But at these low to moderate doses, the initiated injury does not expand to cause liver failure and death of the animals due to the stimulated liver regeneration. At lethal doses, however, the initiated liver injury expands continuously causing liver failure and death of the animals. 1 Interestingly, all animals survive even if a lethal dose is administered 12 to 36 hours after a low-dose priming treatment with another toxicant. These phenomena are known as autoprotection when both the toxicants are same (A + A) or heteroprotection for different toxicants (A + B). 1 -4
Previous work revealed that sustained compensatory tissue repair (CTR), induced by a priming dose of a toxicant, underlies the autoprotection and heteroprotection phenomena. 2 -6 When cell division was inhibited using colchicine intervention, the priming dose was no longer able to protect the animals against a lethal dose treatment. 2 This implies that sustained liver regenerative response, in the form of CTR, after a priming dose treatment confers resiliency against a lethal dose poisoning. Since then, attempts have been made to identify the molecular and cellular mechanisms that determine the timely inception of liver regeneration after toxic liver injury. 7 A few recent studies examined such mechanisms underlying the tightly regulated compensatory liver regeneration. 8 -10 However, the precise mechanisms that can act as a flexible switch to effectively mount and timely terminate compensatory liver regeneration after toxic liver injury have not been understood so far. This necessitates the investigation of such mechanisms to understand the toxicodynamics of the autoprotection and heteroprotection models of toxicity.
Several studies have examined the underlying machineries for tissue regeneration after toxicant-induced and traumatic organ injuries (liver, kidney, lung, heart, and axon). 7,11 -16 Of these cytokine-, chemokine-, growth factor-, neuroendocrine-, mitogenic pathway-, and stem cell-based mechanisms of various organs regeneration, canonical Wnt/β-catenin signaling pathway appears a highly promising one for liver regeneration due to the following compelling evidences: (1) of the several hepatic mitogenic pathways studied, only Wnt/β-catenin pathway was directly correlated with liver regeneration after the APAP treatment 17 ; (2) stimulation of Wnt/β-catenin pathway using a synthetic compound tanshinone IIA stimulated rat hepatic oval cells proliferation, 18 which often contributes toward liver regeneration 19 ; (3) prior treatment with a Wnt/β-catenin agonist reversed the inhibited hepatocyte proliferation in the small-for-size liver graft model in Sprague-Dawley rats 20 ; and (4) Wnt/β-catenin pathway can also protect the liver against oxidative injury by preserving mitochondrial functions. 21 These observations highlight the need to further examine the role of the hepatic Wnt/β-catenin signaling to better understand the toxicodynamics of autoprotection and heteroprotection models of hepatotoxicity.
In addition to the stimulation of CTR, modulation of injury pathways could also reveal the molecular mechanisms of autoprotection and heteroprotection of hepatotoxicity. Among several known pathways for liver injury, the one that can distinctly regulate both hepatocyte proliferation and liver injury holds prime importance. The c-Jun-N-terminal kinase (JNK) pathway is one such pathway with the distinct dual effects. 22 -24 Moreover, the role of JNK pathway in autoprotection and heteroprotection models of toxicity has not been examined so far. This prompted us to study the role of JNK signaling as well in the autoprotection and heteroprotection models of hepatotoxicity.
In our study, we used TA:APAP heteroprotection model, where mice were primed with a small dose of TA or vehicle distilled water (DW) 24 hours before overdosing them with a lethal dose of APAP. After analyzing several pro-regenerative pathways, we found that timely activation of hepatic Wnt/β-catenin signaling and modulation of the JNK pathway drive the heteroprotection by TA against a lethal dose of APAP.
Materials and Methods
Animals
Male Swiss Webster mice (25-30 g), procured from Harlan Laboratories (Indianapolis, Indiana), were used for our studies. The animals were acclimatized in our central animal facility for 5 days before their use in the experiments. The mice were housed over sawdust bedding (Sani-Chips), procured from the Harlan Teklad (Madison, Wisconsin), in air-conditioned facility (21 ± 1°C) under the standard 12-hour light–dark cycle. The animals had unlimited access to rodent chow (Rat Chow No 7001), procured from the Harlan Teklad, and water. All animals were housed and handled as approved by our Institutional Animal Care and Use Committee at the University of Louisiana at Monroe.
Chemicals
Unless otherwise specified, all chemicals and biochemicals were obtained from Sigma-Aldrich (St. Louis, Missouri). For Western blot (WB) analysis, the details of antibodies (supplier, dilution and incubation period) have been provided in the Supplementary Data file. For immunohistochemistry (IHC) staining, the biotinylated secondary antibodies were purchased from Jackson Immunoresearch (West Grove, Pennsylvania).
Treatments
As represented in Figure 1, mice were randomized into 3 groups after 5 days of acclimation period. Group 1 mice received a single dose of APAP vehicle 0.45% of NaCl (20 mL/kg body weight [BW], intraperitoneal [IP]) at 24 hours after TA priming. Group 2 and group 3 mice were primed with vehicle DW (10 mL/kg BW, IP) or a priming dose of TA (40 mg/kg BW, IP) 24 hours before poisoning them with a lethal dose of APAP (600 mg/kg BW, IP). Four mice were euthanized at 1, 6, and 24 hours after the second treatment (ie, vehicle 0.45% of NaCl for group 1 and APAP overdose for group 2 and group 3) in all the groups. Thioacetamide-primed mice euthanized at 24 hours without receiving any second treatment served as a control for the entire study. Four mice from each group were observed for 14 days to record survival and mortality. The administered volumes of vehicles DW and 0.45% of NaCl were same as the administered volumes of respective drugs TA and APAP.

Grouping of the mice and treatment schedule for the thioacetamide (TA): acetaminophen (APAP) heteroprotection study. Note that the second treatment was given at 24 hours after TA (for groups 1 and 3) or distilled water (DW; for group 2) priming. Thioacetamide-primed mice euthanized at 24 hours without giving any treatment served as a control for the entire study. The administered volumes of vehicles DW and 0.45% of NaCl were same as the administered volumes of drugs TA and APAP, respectively.
Biochemical Analysis
Blood samples were collected from the retro-orbital sinus of the mice under light diethyl ether anesthesia. Plasma was collected by centrifugation of blood samples (5000 g, 4°C, 7 minutes) for the biochemical assessments. Entire livers were excised from the mice and immediately frozen in liquid nitrogen before storing them at −80°C for further analysis. Plasma alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities were measured using commercially available kits from Pointe Scientific Co (Canton, Michigan). Protein content in liver homogenate was measured using the commercially available kit from Thermo Scientific Co (Rockford, Illinois).
Histology and IHC
Histology and IHC were performed as described previously. 25
Western Blot Analysis
Western blot analysis of the hepatic proteins was performed using pooled samples as described previously. 26
Real-Time Polymerase Chain Reaction (RT PCR)
Total RNA was isolated from the frozen livers using the TRIzol method according to the supplier’s protocol (Sigma-Aldrich, St. Louis, Missouri). Complementary DNA was prepared from the isolated RNA as described previously. 27 Messenger RNA (mRNA) levels of various genes were determined by SYBR Green-based real-time polymerase chain reaction (PCR) using the Applied Biosystems’ Prism 7300 real-time PCR instrument according to the manufacturer’s protocol (Life Technologies, previously known as Applied Biosystems, Carlsbad, California). S18 gene expression was used as internal standard, and data were expressed as fold change in mRNA expression.
Statistical Analysis
Data are expressed as mean ± standard error of the mean. For multiple comparisons, data were analyzed using 1-way analysis of variance test. Tukey test was used for the post hoc analysis. P ≤ .05 was considered as a statistically significant difference. GraphPad Prism software (GraphPad Software, Inc, La Jolla, California) was used for data analysis.
Results
Plasma ALT and AST Activities
Plasma ALT and AST activities were measured as biomarkers of liver injury at 1, 6, and 24 hours after a lethal dose of APAP to the DW-primed and TA-primed mice. Thioacetamide priming did not affect the rise in ALT (Figure 2A) and AST (Figure 2B) activities seen at 1 hour after a lethal dose of APAP compared to DW + APAP-treated mice. There was a continuous rise in ALT and AST activities after a lethal dose of APAP to the DW-primed mice. In contrast, ALT activity was successively higher at all 3 time points (1, 6, and 24 hours), but AST activity declined at 24 hours compared to the 6-hour time point after a lethal dose of APAP to the TA-primed mice. Notably, while ALT and AST activities were higher at 1 hour, they were significantly lower at 6 and 24 hours in TA + APAP-treated mice compared to DW + APAP-treated mice. There was a rise in both plasma ALT (Figure 2A) and AST (Figure 2B) activities in TA-primed mice measured at 1 hour after the NaCl treatment. Alanine aminotransferase and AST activities peaked at 6 hours and declined thereafter NaCl treatment to the TA-primed mice. These data indicate that, (1) in spite of higher liver injury at 1 hour after APAP overdose to the TA-primed mice compared to that in DW-primed mice, the injury was significantly lower at 6 and 24 hours leading to 100% survival of these mice compared to successively higher injury causing 100% mortality in DW + APAP-treated mice (Table 1), (2) the regression of liver injury in TA + APAP-treated mice had already commenced at 24 hours as reflected by the declined activity of AST whose half-life is much lower than ALT, and (3) priming dose of TA led to minimal and reversible liver injury. All animals, treated with DW + APAP, died at 36 hours after APAP overdose. No mortality was observed until 24 hours after APAP poisoning in the DW + APAP group.

Measurement of plasma (A) alanine aminotransferase (ALT) and (B) aspartate aminotransferase (AST) activities and (C) histopathological examination of the livers at 1, 6, and 24 hours after (1) vehicle 0.45% of NaCl (20 mL/kg body weight [BW]) treatment given at 24 hours in the thioacetamide (TA)-primed mice, that is, TA24 + 1, TA24 + 6, and TA24 + 24, respectively, (2) a lethal dose of acetaminophen (APAP) administered at 24 hours in the distilled water (DW)-primed mice, that is, DW24 + APAP1, DW24 + APAP6, and DW24 + APAP24, respectively, and (3) a lethal dose of APAP administered at 24 hours in the TA-primed mice, that is, TA24 + APAP1, TA24 + APAP6, and TA24 + APAP24, respectively. Results for (A) and (B) are expressed as mean ± standard error of the mean (SEM; n = 4) for each group. *Significantly different (P ≤ .05) from control (*a) or respective DW24 + APAP treatment (*b). C, Arrow and arrowhead represent the necrotic cells and periportal glycogen deposition, respectively. Note that in spite of higher liver injury at 1 hour after APAP overdose in TA-primed mice compared to DW24 + APAP group, the APAP-induced injury did not expand in case of TA-primed mice as indicated by minimal necrosis at 24 hours after a lethal dose of APAP in TA-primed mice (TA24 + APAP24) compared to massive liver necrosis in the DW-primed APAP-overdosed (DW24 + APAP24) mice.
Effect of Various Treatments on the Survival of Mice.a

Abbreviations: APAP, acetaminophen; DW, distilled water; TA, thioacetamide.
aNote that TA priming protected all the mice against APAP overdose-induced lethality. Animals treated with DW + APAP died at 36 hours after APAP overdose. No mortality was observed until 24 hours after APAP poisoning in the DW + APAP group.
Liver Histopathology
The hallmark of APAP-induced liver injury is centrilobular liver necrosis. 3 Classic centrilobular liver necrosis was observed at 1 hour after a lethal dose of APAP to the TA-primed mice compared to nearly healthy appearing centrilobular hepatocytes in the DW + APAP-treated mice (Figure 2C). Only cell swelling was observed in the centrilobular area at 1 hour after a lethal dose of APAP to the DW-primed mice (Figure 2C). In spite of the classic centrilobular necrosis at 1 hour in TA + APAP group, it remained minimal thereafter compared to severely diffused and progressively increased centrilobular necrosis in the DW + APAP-treated mice at 6 and 24 hours after APAP overdose (Figure 2C). Already commenced resolution phase of liver injury in TA + APAP-treated mice was observed at 24 hours as evidenced by liquefaction and minimal number of inflammatory cells in the centrilobular area to aid tissue repair. Priming dose of TA led to periportal glycogen deposition with very minimal centrilobular necrosis at 1 hour and minimal inflammatory response attendant to classic centrilobular necrosis at 6 and 24 hours after the NaCl treatment to these mice (Figure 2C). These data indicated continuous expansion of liver injury after APAP overdose in DW-primed mice causing 100% mortality of these mice. In stark contrast, APAP overdose to the TA-primed mice caused higher liver injury at 1 hour compared to DW + APAP-treated mice, but the initiated injury did not expand beyond 24 hours in the TA + APAP-treated mice as evidenced by the ongoing regression phase of injury at 24 hours and 100% survival of these mice. These histopathological data were in agreement with the biochemical parameters (ALT and AST) of liver injury (Figure 2A and 2B).
These data indicate that (1) priming dose of TA leads to minimal and reversible liver injury yielding 100% survival, (2) lethal dose of APAP to the DW-primed mice leads to unrestrained expansion of liver injury causing 100% mortality (Table 1), (3) priming dose of TA does not interfere with the initiation of APAP-induced liver injury as indicated by the higher liver injury at 1 hour in the TA + APAP-treated mice compared to DW + APAP-treated mice, and (4) in spite of initial higher liver injury in the TA + APAP-treated mice, it regressed thereafter yielding 100% survival of these mice (Table 1). These findings are consistent with previously reported heteroprotection study in rats. 3
Hepatic Expression of Core Cell Cycle Regulatory Proteins
Liver repair in the form of compensatory cell proliferation is stimulated after acute toxic injury to recover the lost tissue. 28 We examined CTR by performing WB analysis for the hepatic core cell cycle proteins such as cyclin D1, cyclin-dependent kinase 4 (CDK4), p21, phosphorylated retinoblastoma (pRb), and proliferating cell nuclear antigen (PCNA) at 1, 6, and 24 hours after APAP overdose to the DW- and TA-primed mice (Figure 3A and 3B). As expected, APAP overdose to the TA-primed mice (TA + APAP) was accompanied by the timely overexpression of cyclin D1, CDK4, and pRb at all time points compared to minimal to no expression of these proteins in the DW + APAP-treated mice (Figure 3A and 3B). Overexpression of cyclin D1, CDK4, and pRb led to timely stimulation of liver regeneration in TA + APAP-treated mice as indicated by quick and sustained overexpression of PCNA protein in these mice compared to very late and minimal expression of PCNA in the DW + APAP-treated mice (Figure 3A and 3B). Unexpectedly, we found overexpression of p21 at 6 hours in TA + APAP-treated mice compared to the DW + APAP-treated mice. Priming dose of TA also led to hepatic cell proliferation as indicated by the overexpression of cyclin D1, CDK4, pRb, and PCNA at 1, 6, and 24 hours after NaCl treatment to the TA-primed mice (Figure 3A and 3B). Expression of p21 protein was not altered significantly in the TA-primed mice (Figure 3A and 3B).
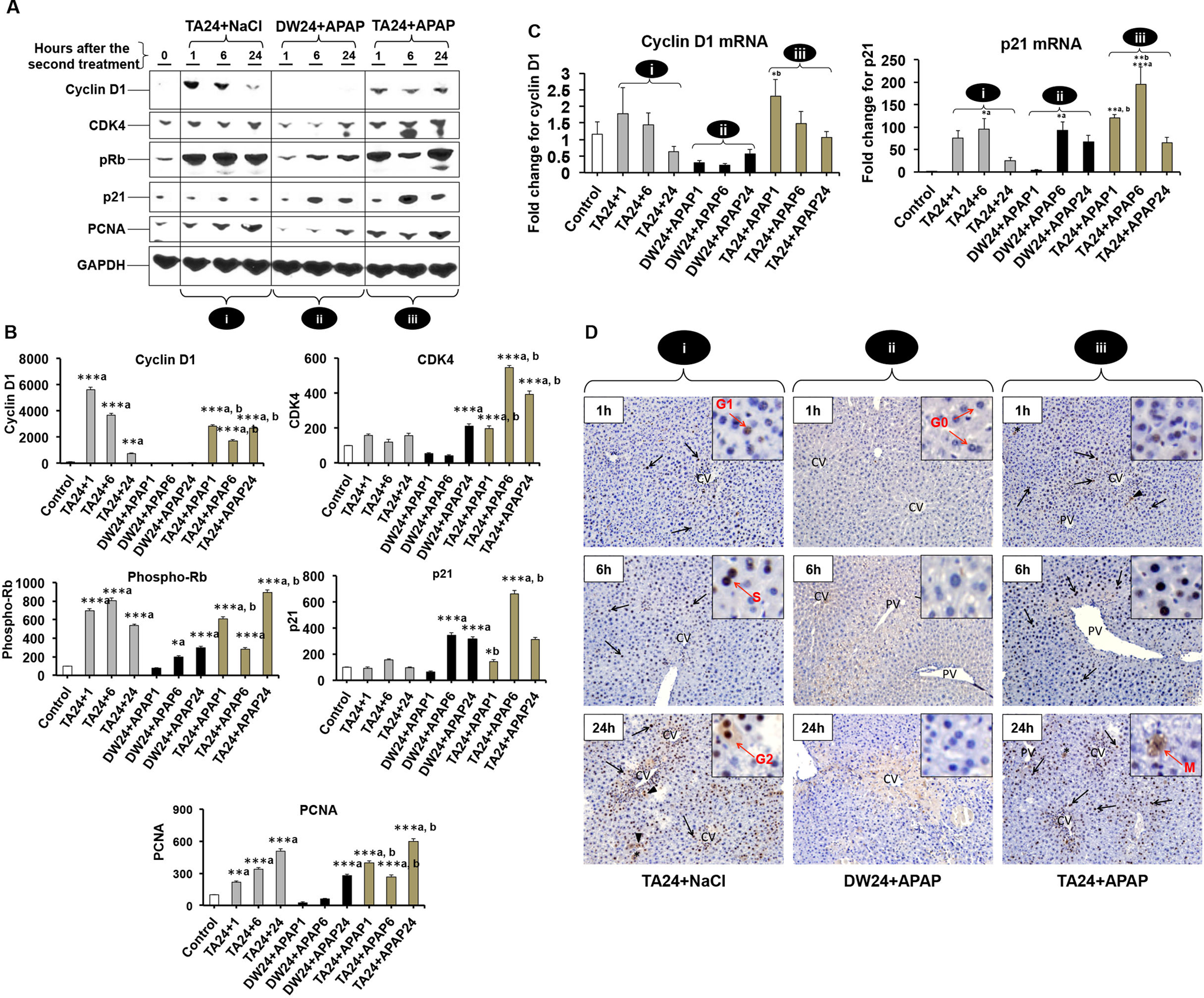
(A) Representative Western blot images for hepatic cyclin D1, cyclin-dependent kinase 4 (CDK4), phosphorylated retinoblastoma (pRb), p21 and proliferating cell nuclear antigen (PCNA) expression at 1, 6, and 24 hours in the (1) TA24 + NaCl-, (2) DW24 + APAP-, and (3) TA24 + APAP-treated mice with (B) respective densitometric analysis. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as an internal control. Note that the expression of cyclin D1, CDK4, and pRb correlated with the PCNA expression. Overexpression of PCNA represents the newly divided or dividing cells (compensatory tissue repair [CTR]). Surprisingly, hepatic expression of p21 was higher in the regenerating livers of the TA24 + APAP mice compared to nonregenerating livers of the DW24 + APAP mice. The justification has been provided in the Discussion section. (C) Measurement of hepatic mRNA expression for cyclin D1 and p21 at 1, 6, and 24 hours in the TA24 + NaCl-, DW24 + APAP-, and TA24 + APAP-treated mice. Results of (B) and (C) are expressed as mean ± SEM (n = 3 for B and n = 4 for C) for each group. *Significantly different (P ≤ .05) from control (*a) or respective DW24 + APAP treatment (*b). (D) Representative photomicrographs of hepatic PCNA IHC analysis at 1, 6, and 24 hours in the TA24 + NaCl-, DW24 + APAP-, and TA24 + APAP-treated mice. Cells in the resting (G0), G1, and S phases show blue, light brown, and dark brown nuclear staining, respectively. Arrowhead and star indicate cells in G2 (diffused brown cytoplasmic staining with brown nuclear staining) and M phases (brown cytoplasmic staining with blue nuclear staining) of cell division, respectively. Arrow, arrowhead, and star indicate PCNA-positive cells. Note the very minimal PCNA-positive staining in DW24 + APAP-treated mice at all the time points examined versus strong and progressively increased PCNA-positive hepatocytes in TA24 + APAP group. Proliferating cell nuclear antigen immunostaining correlated with the hepatic expression of the core cell cycle regulatory proteins (A and B). APAP indicates acetaminophen; DW, distilled water; IHC, immunohistochemistry; mRNA, messenger RNA; TA, thioacetamide.
Collectively, these data indicate that priming dose of TA enabled liver to timely stimulate cell proliferation to mount robust liver repair that eventually led to the regression of injury and 100% survival of the TA + APAP-treated mice (Table 1). In contrast, the delayed and suppressed cell proliferation in DW + APAP-treated mice led to continuous expansion of liver injury and 100% mortality of these mice (Table 1).
Hepatic mRNA Levels of Cyclin D1 and p21
The RT PCR was performed to examine the hepatic cyclin D1 and p21 mRNA levels. Acetaminophen overdose to the TA-primed mice caused a significant increase in the cyclin D1 mRNA immediately at 1 hour compared to the DW + APAP-treated mice. Cyclin D1 mRNA level returned to its normal level at 24 hours in the DW + APAP-treated mice compared to control. Cyclin D1 mRNA level was not altered significantly after a priming dose of TA (Figure 3C). The RT PCR data for cyclin D1 indicate that TA priming enabled APAP-overdosed liver to induce cyclin D1 transcription compared to persistently suppressed transcription in the DW + APAP-treated mice.
Significant induction of p21 mRNA was observed at 6 hours in DW + APAP-treated mice compared to control. Unexpectedly, lethal dose of APAP to the TA-primed mice (TA + APAP) led to a significant increase in the p21 mRNA levels at 1 and 6 hours compared to the DW + APAP-treated mice (Figure 3C). Similarly, there was a significant induction of p21 mRNA in the TA-primed mice at 6 hours after the NaCl treatment (Figure 3C). Unexpected increase in p21 mRNA (Figure 3C) and protein (Figure 3A and 3B) in TA + APAP is supported by the fact that although p21 is conventionally perceived as a negative regulator of cell cycle, it can also promote cell proliferation depending upon the extent of injury. 29,30
Immunohistochemistry Analysis for Hepatic PCNA
The PCNA IHC analysis was performed to qualitatively examine the newly divided or dividing hepatocytes and monitor the progression of cell cycle through 1, 6, and 24 hours after APAP overdose in the DW- and TA-primed mice. Very minimal or no PCNA staining was observed at all time points in DW + APAP-treated mice compared to strong and progressively increased staining in the TA + APAP-treated mice (Figure 3D). Hepatocytes in different stages of cell cycle were identified using this technique. Almost all hepatocytes were in the resting phase (G0) at 1 hour after APAP overdose to the DW-primed mice compared to several hepatocytes in S phase of cell division in the TA + APAP-treated mice. Small number of cells in G2 phase was also found in TA + APAP-treated mice at 1 hour after APAP overdose indicating early onset of cell cycle progression before receiving APAP overdose. Most of the cells remained in G0 phase even at 6 and 24 hours, except a few cells in G1 phase at 24 hours, after APAP overdose to the DW-primed mice. In contrast, a large number of hepatocytes were found in S phase and M phase of cell division at 6 and 24 hours, respectively, after APAP overdose to the TA-primed mice. A few hepatocytes in G1 and S phases of cell division were also found at 24 hours in the TA + APAP treated mice. Qualitatively, PCNA staining in DW + APAP- and TA + APAP-treated mice was consistent with the quantitative analysis of the hepatic PCNA as carried out by WB analysis (Figure 3A and 3B). The priming dose of TA also led to the stimulation of hepatic cell cycle as indicated by the progressive increase in PCNA staining from 1 through 24 hours after NaCl treatment to these mice (Figure 3D). Several hepatocytes in the G1 and S phases of cell division were observed at 1 and 6 hours. Mixed population of cells in S, G2, and M phases were observed at 24 hours after NaCl treatment to the TA-primed mice (Figure 3D).
The PCNA IHC data indicate that priming dose of TA led to the stimulation of cell cycle as a compensatory mechanism to replace the dying cells and overcome the initiated injury. Moreover, TA priming enabled liver to mount robust cell proliferation even after a lethal dose of APAP to these mice compared to inhibited cell proliferation in the DW + APAP-treated mice.
Hepatic Expression of β-Catenin Pathway Proteins
In order to examine the role of β-catenin pathway in liver regeneration, WB analysis was performed for hepatic total β-catenin, inactive species of β-catenin, represented by the phosphorylation of β-catenin at S45/Thr41 and S33/37/Thr41 sites 31 and active β-catenin. Acetaminophen overdose to the DW-primed mice led to a significant decrease in total β-catenin at 1 hour after APAP overdose compared to control mice (Figure 4A and 4B). Hepatic total β-catenin was preserved at all time points (1, 6, and 24 hours) in TA + APAP-treated mice compared to control (Figure 4A and 4B). Total β-catenin in the TA + APAP-treated mice at 6 hours was significantly higher than the DW + APAP-treated mice. Priming dose of TA also led to overexpression of total β-catenin as seen at 1 hour after the NaCl treatment (Figure 4A and 4B). We then analyzed the inactive forms of β-catenin (phospho-β-catenin). Significant increase in phosphorylation of β-catenin was observed over 24 hours after APAP overdose to the DW-primed mice compared to control (Figure 4A and 4B). Although one species of phospho-β-catenin (S45/Thr41) was significantly overexpressed immediately at 1 hour, the other species of the inactive β-catenin (S33/37/Thr41) was significantly overexpressed at 6 hours and lasted until 24 hours in the DW + APAP-treated mice compared to control (Figure 4A and 4B). There was an overlap between the overexpression of both species of the phosphorylated β-catenin at 6 hours in the DW + APAP-treated mice compared to control. In contrast, expression of the neither species of phosphorylated β-catenin (inactive forms) was altered until 24 hours after the APAP overdose to the TA-primed mice compared to control (Figure 4A and 4B). Moreover, significant overexpression of both species of the phospho-β-catenin was observed late at 24 hours in TA + APAP-treated mice compared to DW + APAP-treated mice and controls. Priming dose of TA also led to significant overexpression of 1 species (S33/37/Thr41) of phospho-β-catenin (Figure 4A and 4B).

(A) Representative Western blot images for hepatic β-catenin, phospho-β-catenin (S45/Thr41 and S33/37/Thr41), and active β-catenin expression at 1, 6, and 24 hours in the TA24 + NaCl-, DW24 + APAP-, and TA24 + APAP-treated mice with (B) respective densitometric analysis. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as an internal control. Overexpression of total and active β-catenin was observed in TA24 + APAP mice compared to very minimal to negligible expression in DW24 + APAP mice. Note the marked overexpression of phospho-β-catenin after a lethal dose of APAP in DW-primed mice (DW24 + APAP) compared to TA24 + APAP mice. (C) Measurement of hepatic mRNA expression for axin2 at 1, 6, and 24 hours in the TA24 + NaCl-, DW24 + APAP-, and TA24 + APAP-treated mice. Results of (B) and (C) are expressed as mean ± SEM (n = 3) for each group. *Significantly different (P ≤ .05) from control (*a) or respective DW24 + APAP treatment (*b). Notably, axin2 mRNA level was significantly higher at 1 and 6 hours in TA24 + APAP-treated mice compared to the DW24 + APAP-treated mice. APAP indicates acetaminophen; DW, distilled water; mRNA, messenger RNA; TA, thioacetamide.
We then examined the expression of active form of β-catenin to further confirm the activation of β-catenin pathway in the liver regeneration. Although there was no change in the expression of active β-catenin in the DW + APAP-treated mice, the TA + APAP-treated mice showed a significant overexpression of active β-catenin at 6 and 24 hours after APAP overdose compared to the DW + APAP and control mice (Figure 4A and 4B). Priming dose of TA did not alter the expression of active β-catenin seen at 1, 6, and 24 hours after NaCl treatment to these mice (Figure 4A and 4B).
Notably, the activation of β-catenin pathway in TA + APAP-treated mice was consistent with the differential expression of phosphorylated species and activated β-catenin in these mice compared to DW + APAP-treated mice (Figure 4A and 4B). Collectively, these data indicate that there was timely activation of β-catenin pathway in TA + APAP compared to its persistent lack in the DW + APAP-treated mice.
Hepatic mRNA Levels of Axin2
Axin2 is a direct target gene of β-catenin pathway with negative feedback mechanism on the β-catenin signaling. 32,33 In our study, APAP overdose to the DW-primed mice led to a significant decrease (5-fold) in hepatic Axin2 mRNA at 6 hours compared to the control mice (Figure 4C). In contrast, APAP overdose to the TA-primed mice led to 2-fold increase in hepatic Axin2 mRNA at 6 hours compared to control mice (Figure 4C). Hepatic Axin2 mRNA level in TA + APAP-treated mice was significantly higher by 2.5-fold and 9.7-fold at 1 and 6 hours, respectively, compared to the DW + APAP-treated mice (Figure 4C). Priming dose of TA did not significantly alter the Axin2 mRNA level seen at 1, 6, and 24 hours after 0.45% NaCl treatment to these mice compared to control (Figure 4C). These data are in agreement with the early activation of β-catenin pathway in TA + APAP-treated mice compared to DW + APAP-treated mice.
Hepatic Total JNK, Phospho-JNK, and Cyp2e1 Proteins
In order to examine the attendant signaling of APAP-induced liver injury, we chose to examine hepatic total JNK and phospho-JNK proteins in the DW + APAP- and TA + APAP-treated mice. Acetaminophen overdose led to significant overexpression of total JNK in both the DW + APAP- and TA + APAP-treated mice at 6 and 24 hours compared to control mice (Figure 5A and 5B). Hepatic total JNK was significantly higher at 1 and 6 hours in the TA + APAP-treated mice compared to the DW + APAP-treated mice (Figure 5A and 5B). Priming dose of TA also led to significant overexpression of total JNK (Figure 5A and 5B). To examine the effect of priming treatments on JNK activation, hepatic phospho-JNK (activated form) was measured in the DW + APAP- and TA + APAP-treated mice. Phospho-JNK was moderately overexpressed at 1 hour and thereafter in the TA + APAP-treated mice compared to its late but remarkably high expression at only 6 hours in the DW + APAP-treated mice (Figure 5A and 5B). Notably, expression of phospho-JNK at 1 hour in the TA + APAP-treated mice was significantly higher than control mice. Priming dose of TA also led to significant overexpression of phospho-JNK (Figure 5A and 5B).

(A) Representative Western blot images for hepatic total c-Jun N-terminal kinases (JNK), phospho-JNK, and Cyp2e1 expression at 1, 6, and 24 hours in the TA24 + NaCl-, DW24 + APAP-, and TA24 + APAP-treated mice with (B) respective densitometric analysis. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as an internal control. Results of (B) are expressed as mean ± SEM (n = 3) for each group. *Significantly different (P ≤ .05) from control (*a) or respective DW24 + APAP treatment (*b). Notably, moderate but quick and sustained overexpression of phospho-JNK was observed at 1 hour in TA24 + APAP mice compared to late and remarkable high expression at 6 hours in DW24 + APAP mice. APAP indicates acetaminophen; DW, distilled water; TA, thioacetamide.
Finally, we examined hepatic Cyp2e1 protein level after all 3 treatments. Both, TA and APAP are bioactivated through hepatic Cyp2e1 into reactive toxic metabolites to initiate liver injury. Although there was no change in hepatic Cyp2e1 until 24 hours after TA priming (served as control group), its level significantly decreased thereafter (Figure 5A and 5B). After confirming that TA priming would not influence the hepatic Cyp2e1, we administered a lethal dose of APAP at 24 hours after TA priming in our heteroprotection study.
Discussion
It is known that living organisms are equipped with several endogenous defense mechanisms against physical, chemical, and biological offending agents. 34 -36 The CTR is one such mechanism that can even rescue the toxicant-overdosed death-bound animals. 36 Infliction of tissue injury at sublethal doses of toxicants stimulates CTR in order to overcome the tissue damage. 1 Extensive research over the last several decades has revealed that CTR plays a crucial role in determining the fate of the initiated injury. 1,36 -44 Quick onset of adequate CTR, as in the case of low to moderate dose of toxicants, leads to the regression of injury. 45,46 On the other hand, lethal dose treatment causes suppression of the CTR giving a freeway for the unabated expansion of the initiated injury. Unrestrained injury progression leads to the organ failure eventually causing death of the animals. 45,46
Given this deterministic role of CTR to regulate the fate of toxic tissue injury, it is imperative to identify the underlying molecular mechanisms driving CTR. Upon infliction of injury at low to moderate doses, the dying cells trigger a cascade of distress signals by releasing several cytokines and growth factors. 47 -50 These signals eventually stimulate the surrounding healthy or minimally affected cells to undergo rapid proliferation. These mechanisms have been accepted for the stimulation of compensatory cell proliferation. However, the initiated compensatory cell proliferation should end precisely to avoid uncontrolled cell division and its progression to unwanted tissue growth. Molecular signaling that can act as a flexible switch for timely onset and appropriate termination of the compensatory liver regeneration has not been identified so far.
In this study, we used TA:APAP heteroprotection model to examine the mechanisms underlying CTR-mediated heteroprotection against APAP-induced lethal liver injury. We stimulated CTR by a priming dose of TA (40 mg/kg) administered at 24 hours before poisoning the animals with a lethal dose of APAP. This priming dose of TA is one-twelfth of the LD100 dose in mice. 51 As expected, priming dose of TA completely protected mice against APAP overdose-induced liver injury (Figure 2) by timely mounting CTR (Figure 3). In stark contrast, the inhibited CTR in the DW + APAP-treated mice led to the continuous expansion of liver injury causing 100% mortality of these mice (Table 1). Surprisingly, hepatic p21 protein (Figure 3A and 3B) and mRNA levels (Figure 3C) were also higher at the peak of DNA synthesis in the regenerating livers of the TA + APAP-treated mice compared to the DW + APAP-treated mice (Figure 3). Protein p21 has been best defined as negative regulator of cell cycle due to its inhibitory action against cyclin/CDK kinase activity. 52 However, consistent to our findings, it has been reported that p21 is upregulated during liver regeneration depending upon the degree of liver injury and the phase of cell cycle progression. 29,53,54 Time course studies with a multiple fold dose range of mechanistically diverse hepatotoxicants are required to determine the differential role of p21 in the liver regeneration. Our WB analysis of the core cell cycle proteins and PCNA IHC analysis indicate that stimulation of cell cycle at an early stage underlies the heteroprotection by TA against APAP overdose.
After confirming the deterministic role of compensatory liver regeneration in the final outcome of toxic liver injury, we investigated molecular mechanisms underlying the timely onset of CTR. Of the several known regenerative pathways, we examined the role of canonical Wnt/β-catenin signaling pathway in compensatory liver regeneration. Rationale for examining Wnt/β-catenin signaling pathway in our heteroprotection model of hepatotoxicity is that, (1) pro-regenerative cyclin D1 is a target gene for β-catenin signaling pathway 55 and (2) activation of β-catenin signaling through endogenous and pharmacological interventions caused compensatory liver regeneration. 17 -19 Moreover, Wnt/β-catenin was recently shown to exert antioxidant and mitochondrial protective effects during ischemia reperfusion liver injury. 21 In our studies, we found early and robust activation of β-catenin pathway as indicated by overexpression of total and activated β-catenin in the TA + APAP-treated mice compared to the DW + APAP-treated mice (Figure 4A and 4B). Overexpression of total and activated β-catenin during liver regeneration and oval cell proliferation after APAP hepatotoxicity and PH, respectively, has already been reported. 27,56 Increased expression of phospho-β-catenin and decreased expression of activated β-catenin at 24 hours after APAP overdose to the TA-primed mice indicate timely termination of the pro-regenerative signals in the TA + APAP-treated mice. Differential activation of β-catenin pathway in the DW + APAP- and TA + APAP-treated mice was supported by the coinciding mRNA levels of the β-catenin target gene Axin2 in these mice (Figure 4C). Notably, target gene Axin2 acts via negative feedback mechanism to inhibit β-catenin activation. 32 This is consistent with the inhibited β-catenin signaling at 24 hours after APAP overdose in the TA-primed mice. This way, β-catenin signaling acted as a flexible switch that determined timely onset and appropriate termination of the pro-regenerative signals during compensatory liver regeneration in the TA + APAP-treated mice. Based on the differential expression of β-catenin pathway proteins, axin2 mRNA levels, and PCNA IHC data, it can be interpreted that early activation of β-catenin facilitated liver regeneration by promoting G1/S phase cell cycle transition in the TA + APAP-treated mice. The role of β-catenin in facilitating G1/S transition has been reported earlier. 27
Although role of JNK signaling in APAP hepatotoxicity has remained controversial, extent and duration of JNK activation (phospho-JNK expression) determine its role in the APAP hepatotoxicity and compensatory liver regeneration. 22,24,57 Consistent with these reports, we found a remarkable overexpression of phospho-JNK at 6 hours after APAP overdose to the DW-primed mice compared to the TA + APAP-treated mice (Figure 5A and 5B). Given the dual role of JNK activation in mediating liver injury 57,58 and driving liver regeneration by facilitating G0 to G1 transition, 22 its activation at 6 hours, although remarkable, is too late to timely mount robust liver regeneration in the DW + APAP-treated mice (Figure 5A and 5B). Therefore, JNK activation at 6 hours in the DW + APAP-treated mice mediated APAP-induced liver injury. In stark contrast, moderate and timely activation of JNK at 1 hour and thereafter in the TA + APAP-treated mice facilitated G0 to G1 transition to mount robust liver regeneration in these mice. Positive relationship between JNK activation and liver regeneration is also supported by its minimal (but significant compared to control) expression in the TA-primed mice at 1 and 6 hours post saline treatment to these mice (Figure 5A and 5B).
Collectively, our data indicate that hepatic β-catenin pathway promptly acts as a flexible switch to timely mount and appropriately terminate the compensatory liver regeneration after APAP overdose by facilitating the G1/S transition. Moreover, moderate and early activation of JNK signaling assisted in the continuous supply of new G1 cells by facilitating G0/G1 transition. Interestingly, early activation of both JNK and β-catenin is required to timely mount compensatory liver regeneration. This conclusion can be derived based on the fact that in spite of remarkably high JNK activation at 6 hours in the DW + APAP-treated mice, the compensatory liver regeneration was promptly terminated due to inhibited β-catenin signaling in these mice. Therefore, we conclude that complimentary roles of JNK and β-catenin (Figure 6) drive a very well-balanced liver regeneration in TA:APAP heteroprotection model of hepatotoxicity. Timely activation and appropriate termination of the β-catenin signaling were the hallmark of balanced liver regeneration in the TA + APAP-treated mice.
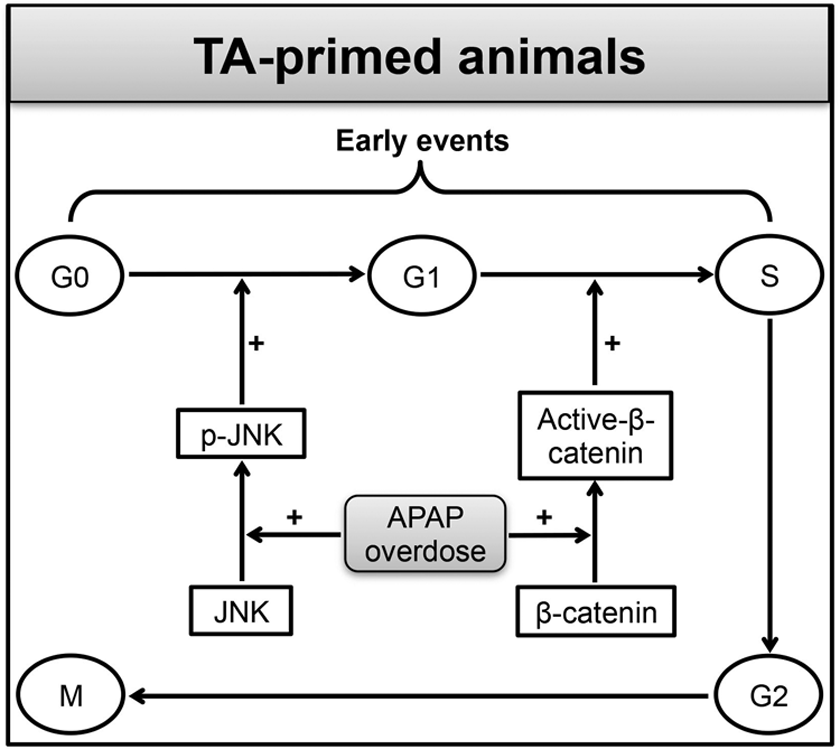
Diagrammatic representation of the molecular mechanisms underlying heteroprotection by thioacetamide (TA) priming against a lethal dose of acetaminophen (APAP). Note that most of the proliferating cell nuclear antigen (PCNA)-stained hepatocytes were in early phase of cell division (G1 phase) at 24 hours after TA priming. Note that early activation of β-catenin after a lethal dose of APAP in the TA-primed mice facilitates G1/S transition to initiate timely compensatory tissue repair (CTR). The activity of β-catenin is complimented by early activation of c-Jun N-terminal kinases (JNK; p-JNK expression) that facilitates G0/G1 transition for continued cell cycle progression. Due to inhibited β-catenin activation and delayed activation of JNK, DW24 + APAP mice fail to timely mount robust liver generation after APAP overdose.
Footnotes
Acknowledgments
The author gratefully appreciates the South Central Chapter of the Society of Toxicology (SOT) for providing with a Technology Transfer Award to visit Dr. Udayan Apte’s laboratory at KUMC, KS, USA, to perform the entire work described here. The author is equally thankful to the Molecular and Systems Biology Specialty Section and Mechanisms Specialty Section of the SOT for recognizing this work for the Graduate Student Research Award (third place) and Carl C. Smith Graduate Student Award (finalist), respectively. Heartfelt gratitude to Dr. Mehendale for the financial support and Dr. Udayan Apte for allowing to perform the entire work in his laboratory at KUMC, KS, USA.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.