
Abstract
Hormesis and adaptive responses are 2 important biological effects of low-dose ionizing radiation (LDR). In normal tissue, LDR induces hormesis as evinced by increased cell proliferation; however, whether LDR also increases tumor cell proliferation needs to be investigated. In this study, cell proliferation was assayed by total cell numbers and the Cell Counting Kit 8 assay. Mitogen-activated protein kinases (MAPK)/extracellular signal-regulated kinase (ERK) and phosphatidylinositol 3′ -kinase(
Introduction
Lung cancer is the leading cause of cancer deaths worldwide with millions of new cases diagnosed every year. 1 Approximately 15% to 20% of lung cancer belongs to the histological subgroup classified as small-cell lung cancer (SCLC). 2 Small-cell lung cancer is a fast-growing neuroendocrine tumor that differs considerably from non–small-cell lung cancer in biology, response to therapy, and final prognosis. It is characterized by an aggressive biological behavior and a distinctive likelihood of systemic spread. 2,3 In general, cancer cells show increased radiosensitivity owing to a breakdown in cell cycle checkpoints and DNA repair mechanisms, and this increased radiosensitivity leads to an accumulation of irreparable DNA lesions and cell death. 4 Accordingly, although SCLC exhibits high sensibility to chemotherapy, radiotherapy also plays an important role in its treatment. 5 However, it is common for patients with cancer to have pain, fatigue, and skin reactions when receiving radiotherapy 6 because of radiation damage to the adjacent normal tissues. 4 Thus, new therapies with fewer side effects are needed to improve the treatment of SCLC.
The induction of adaptive response by low-dose radiation (LDR) in vitro and in vivo has promoted us to explore whether the adaptive response induced by pretreatment with LDR could be applied to cancer therapy. Our previous studies showed that LDR stimulated cell proliferation in normal cells including mesenchymal stem cells, but this might not occur in solid tumor cells. 7,8 If LDR can induce hormesis in normal tissue cells while leaving adjacent tumor cells unaffected, it might be a feasible strategy to provide patients having cancer with LDR exposure prior to radiotherapy to reduce the degree of radiotherapy-induced damage to normal tissues without attenuating its therapeutic effect on tumor cells. 7,8 However, most previous studies have utilized either normal cell lines or tumor cell lines individually, and there have been few studies that have directly compared normal and tumor cells under the same experimental conditions.
In this study, therefore, we examined the role of LDR in human embryonic lung fibroblast 2BS and small lung cancer NCI-H446 cell lines. We also mechanistically examined the roles of the mitogen-activated protein kinases (MAPK)/extracellular signal-regulated kinase (ERK) and phosphatidylinositol 3′ -kinase(
Materials and Methods
Cell Culture and Reagents
Human embryonic lung fibroblasts (2BS cells) were previously isolated from female fetal lung fibroblast tissue and have been fully characterized. 9 Briefly, the 2BS cell line was originally established at the National Institute of Biological Products (Beijing, China) and was maintained in low glucose Dulbecco modified Eagle medium (Gibco, Gaithersburg, Maryland) supplemented with 10% fetal bovine serum, penicillin (100 U/mL), and streptomycin (100 μg/mL). The lung cancer NCI-H446 cell line, purchased from the American Type Culture Collection (Manassas, Virginia), was cultured with the same medium as the 2BS cell line. Cultures were maintained at 37°C in a humidified 5% CO2 incubator. The medium was changed every 3 days. The specific MEK1/2 inhibitor, U0126, and PI3K inhibitor, LY294002, were obtained from Beyotime (Haimen, China).
Irradiation
Exponentially growing cells were irradiated with X-rays (Varian Clinac 21EX, Palo Alto, California) at room temperature. The dose rate for X-irradiation was 100 mGy/min. Control groups were treated similarly except for irradiation. After exposure to LDR, cells were cultured in a 37°C incubator for further experiments.
Proliferation and Live Cell Counting
Cells (5 × 104) were seeded into 60-mm cell culture plates and incubated for 20 hours before irradiation. Then, cells were irradiated with 0, 20, 50, 75, or 100 mGy X-rays. At 24 hours after LDR, cells were digested with trypsin and divided into 2 parts: the first was directly counted and the total cell number calculated, and the remainder was seeded into 96-well culture dishes for the detection of cell proliferation using the Cell Counting Kit 8 (CCK-8) assay. For the latter, 20 μL CCK-8 solution (Biyuntian, Haimen, China) was added into each well and incubated at 37°C with 5% CO2 for 3 to 4 hours. After this additional incubation, the absorbance was measured in a microplate reader (Bio-Rad Laboratories, Berkeley, California) at 450 nm. The experiments were performed in triplicate and repeated for a minimum of 3 times.
Cell Cycle Analysis
Cell cycles were analyzed by flow cytometry, for which the cells (10 000) were harvested at 2, 6, and 24 hours after 50 mGy LDR and fixed with ice-cold 70% ethanol at 4°C overnight. Then, the fixed cells were washed twice with phosphate-buffered saline (PBS) and stained with 50 μg/mL propidium iodide in the presence of RNase A (10 μg/mL). The stained cells were analyzed for DNA content by fluorescence-activated cell sorting in a BD FASCalibur instrument (Becton Dickinson, Bedford, Massachusetts).
Western Blot Analysis
Cells were collected from the 100-mm culture dishes and washed with ice-cold PBS and lysed at 4°C on ice for 20 minutes in 100 μL lysis buffer (50 mmol/L Tris-HCl, pH 7.5; 0.25% deoxycholate; 1% Triton X-100; and 0.5 mol/L NaCl). Protein concentrations were determined using the Bradford method. Equal aliquots of the protein sample (30 μg/lane) were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes. The blot was then incubated for 1 hour with blocking buffer containing 0.1% Tween 20 and 5% nonfat dry milk. The membranes were incubated with primary antibodies overnight (phospho-c-Raf [p-c-Raf], cellular rapidly accelerated fibrosarcoma [c-Raf], phospho-extracellular signal-regulated kinase1/2 [p-ERK1/2], extracellular signal-regulated kinase1/2 [ERK1/2], phospho-MAPK and extracellular signal-regulated kinase1/2 [p-MEK1/2], MAPK and extracellular signal-regulated kinase1/2 [MEK1/2], phospho-3′-phosphoinositide-dependent kinase-1 [PDK-1], phospho-3′-phosphoinositide-dependent kinase-1 [p-PDK-1], AKT, and phospho-AKT [p-AKT], respectively, purchased from Cell Signaling Technology, Danvers, Massachusetts) at a dilution of 1:1000 followed by incubation with the secondary antibody (Santa Cruz Biotechnology, Dallas, Texas). An Enhanced chemiluminescence detection system was used to visualize the proteins.
Statistical Analysis
Data were expressed as means ± standard deviation (SD). Statistical significance among groups was determined using the Student t test. Differences with a P value <.05 were considered significant.
Results
Effects of LDR on Cell Growth and the Cell Cycle in 2BS and NCI-H446 Cell Lines
The effects of LDR on 2BS and NCI-H446 cells were investigated by live cell counting (Figure 1A and C) and cell proliferation assessment using the CCK-8 assay (Figure 1B and D). As shown in Figure 1A and B, the total cell numbers and cell proliferation were significantly enhanced in the 2BS cells upon exposure to LDR at doses of 20 to 75 mGy, and the values peaked at 50 mGy but were not changed compared with controls following exposure to 100 mGy (P < .05). However, under the same experimental conditions, the total NCI-H446 cell numbers and proliferation were not stimulated and even decreased compared to the controls (P < .05, Figure 1C and D).
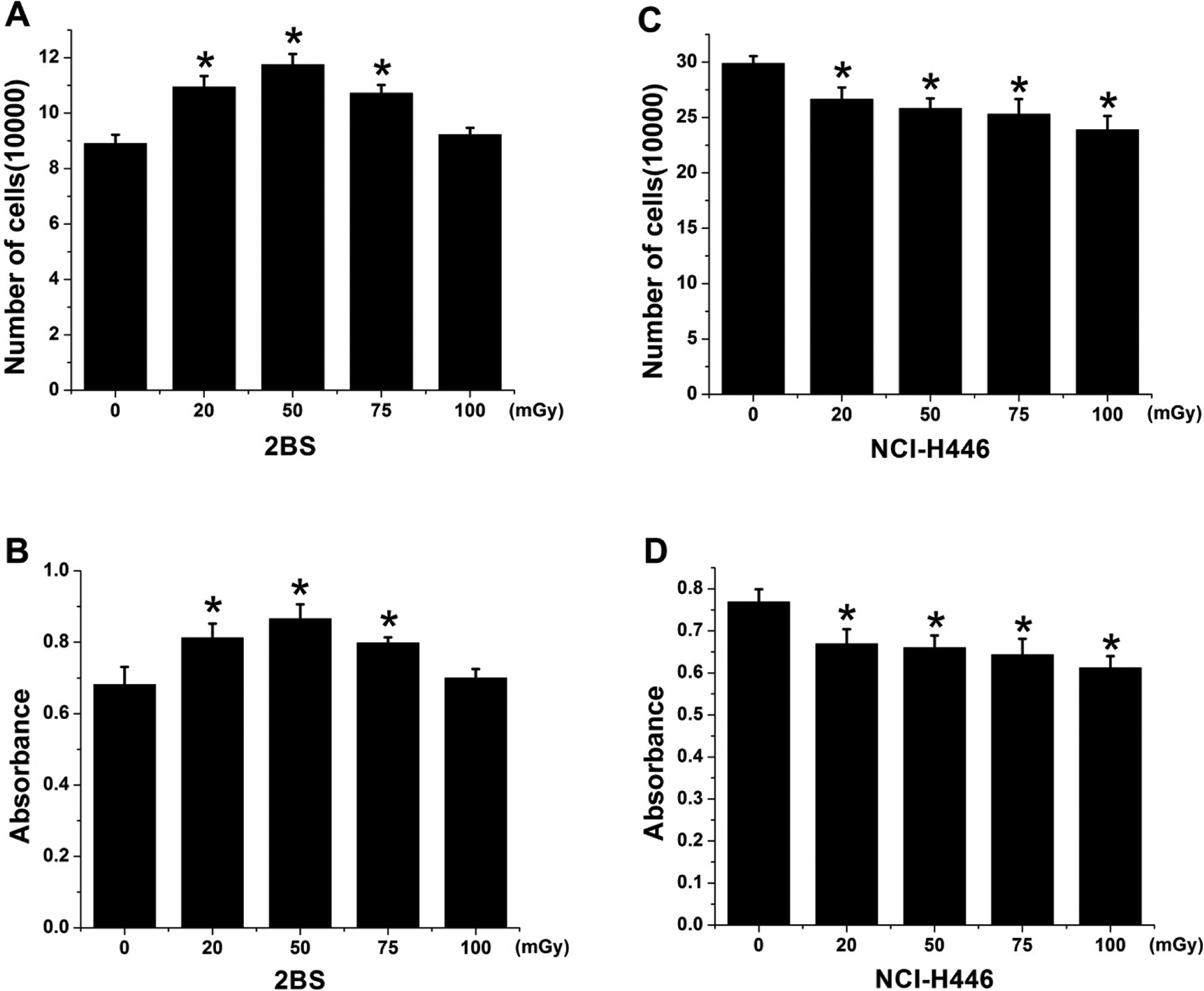
Low-dose ionizing radiation (LDR) enhanced the cell proliferation of 2BS but not NCI-H446 cells. A and C, Cells (5 × 104) were seeded in 60 mm cell culture plates. The number of cells was counted to determine the initial cell number. Then, the cells received various doses of X-rays and were incubated for an additional 24 hours before the cell number was recounted. B and D, Cell proliferation was determined by the Cell Counting Kit 8 (CCK-8) assay. Data are presented as the means ± standard deviation (SD) of 3 separate experiments with duplicate samples at the minimum. *P < .05 vs control.
To determine how radiation affected cell cycle progression, cells were collected at 2, 6, and 24 hours following exposure to 50 mGy X-irradiation and subjected to flow cytometry analyses. The results demonstrated that there was almost a 1- to 4-fold increase in the number of 2BS cells in the S-phase from 2 to 6 hours after LDR. However, there was no change observed in the percentage of NCI-H446 cells in the S-phase of the cell cycle within 24 hours after exposure to LDR (Figure 2A and B). These data suggest that there is a rapid response of 2BS cells to LDR, resulting in a transient progression of cells from G1- to S-phase. In contrast, a significantly time-dependent increase in apoptotic cell death was evident in NCI-H446 cells exposed to 50 mGy X-rays (Figure 2C).

Low-dose ionizing radiation (LDR) enhanced the number of S-phase cells in 2BS but not NCI-H446 cells. A, The profiles of each cell line were analyzed 0, 2, 6, and 24 hours following radiation exposure by flow cytometry using propidium iodide staining for DNA content. The graphs represent the percentage of cells in each phase of the cell cycle. B, The data show increases in the number of S-phase cells and in cells undergoing apoptosis. Data are presented as the means ± standard deviation (SD) of 3 separate experiments with duplicate samples at a minimum. *P < .05 vs control.
Phosphorylation of c-Raf, MEK1/2, and ERK1/2 in 2BS and NCI-H446 Cells
Our previous study had indicated that there was an association between LDR-stimulated cell proliferation in normal rat mesenchymal stem cells in vitro and the activation of the MAPK/ERK pathway. 7 To determine whether any difference in the MAPK/ERK signal pathway in response to LDR could be associated with the different cell proliferation responses observed between 2BS and NCI-H446 cell lines, the phosphorylation of c-Raf, ERK1/2, and MEK1/2 was examined at 6 hours after exposure to different doses of X-irradiation. In 2BS cells, the amount of phosphorylated ERK1/2 increased about 3- to 12-fold from 20 to 75 mGy compared with the control and peaked at 50 mGy. We also observed approximately 2- to 5-fold increases in the phosphorylation of upstream mediators of MEK1/2 and c-Raf in 2BS cells in response to 20 to 75 mGy X-irradiation, which peaked at 50 mGy. In NCI-H446 cells, however, neither the phosphorylated nor the total c-Raf, MEK1/2, or ERK1/2 levels were altered (Figure 3).
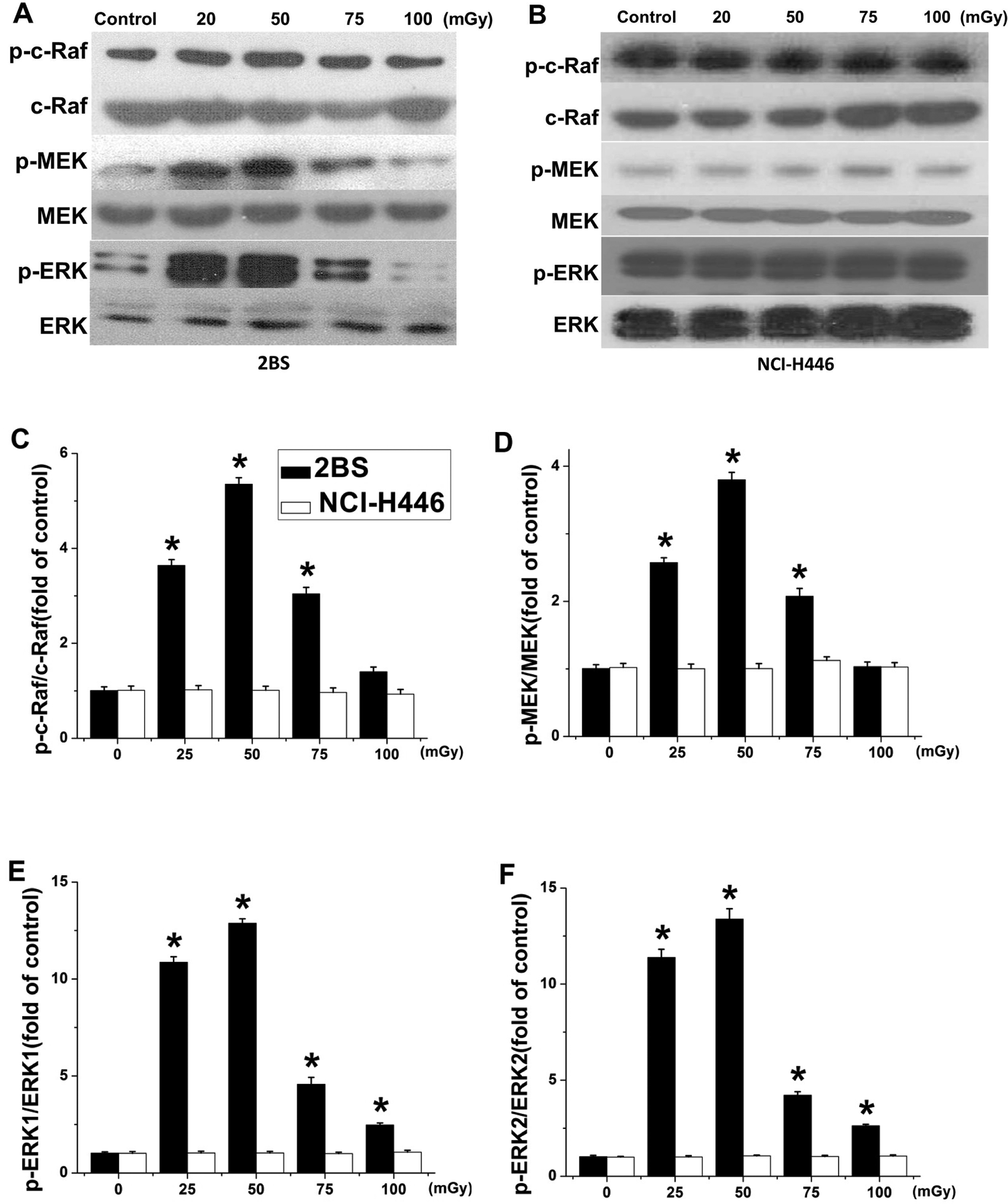
Low-dose ionizing radiation (LDR) induced phosphorylation of c-Raf, MEK1/2, and ERK1/2 in 2BS but not NCI-H446 cells. Phosphorylation of c-Raf ERK1/2 and MEK1/2 was determined 6 hours after exposure to 20, 50, 75, or 100 mGy X-irradiation by Western blot analysis for both 2BS (A) and NCI-H446 (B) cell lines. Quantitative analysis of the phosphorylated to total expression ratio for c-Raf (C), MEK (D), and ERK1/2 (E and F) was determined. Data are presented as means ± standard deviation (SD) of 3 separate experiments with duplicate samples at a minimum. *P < .05 vs control.
To determine whether the activation of the PI3K/AKT signaling pathway is associated with the observed cell proliferation, phosphorylation of PDK-1 and AKT was examined after irradiation. 2BS and NCI-H446 cells were irradiated with 20, 50, 75, and 100 mGy X-rays and incubated for 6 hours. In 2BS cells, the levels of phosphorylated AKT were observed to have increased about 1.5- to 3-fold compared with the controls. The phosphorylation of the upstream effector of AKT, PDK-1, was also increased about 1.7- to 6-fold, but the total levels of PDK-1 and AKT were not changed. In NCI-H446 cells, however, neither the phosphorylated nor the total PDK-1 AKT levels were altered (Figure 4).
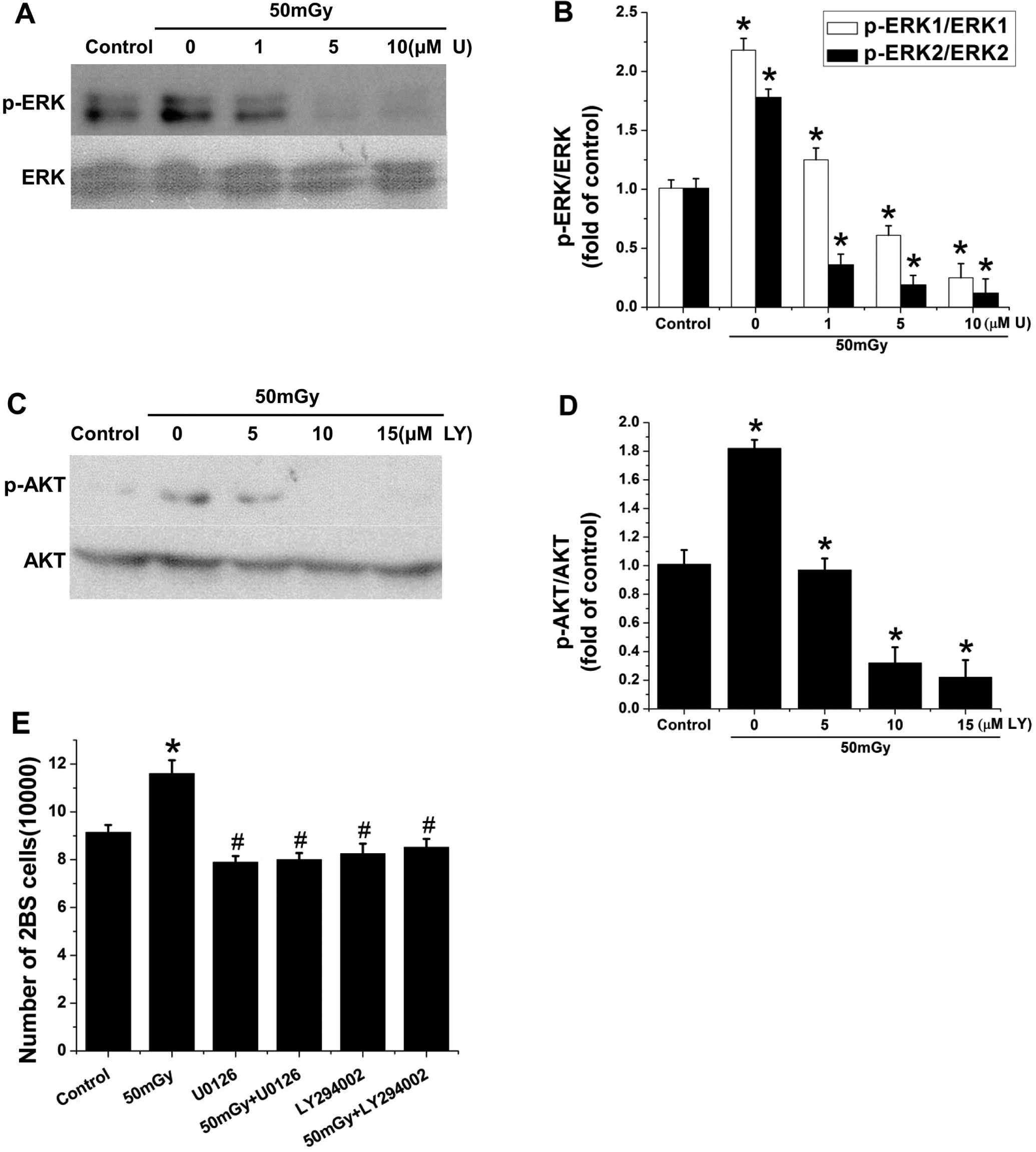
Inhibition of ERK1/2 or AKT prevented low-dose ionizing radiation (LDR)-induced cell proliferation in 2BS cells. A and B, Cells were pretreated with and without 1, 5, or 10 μmol/L U0126 for 1 hour and then irradiated with 50 mGy X-rays. Phosphorylation of ERK1/2 was determined by Western blot and quantitatively analyzed relative to total ERK1/2. C and D, Cells were pretreated with and without 5, 10, and 15 μmol/L LY294002 for 1 hour and then irradiated with 50 mGy X-rays. Phosphorylation of AKT was determined by Western blot analysis, followed by quantitative analysis. E, Exponentially growing cells were pretreated with 10 μmol/L U0126 or 10 μmol/L LY294002 for 1 hour and then were irradiated with 50 mGy X-rays. Cells were incubated for an additional 24 h before counting. Data are presented as the means ± standard deviation (SD) of 3 separate experiments with duplicate samples at a minimum. U: U0126; LY: LY294002. *P < .05 vs control; # P < .05 vs 50 mGy group.
Effect of Inhibitors on ERK1/2 and AKT Phosphorylation and on Enhanced Cell Proliferation in 2BS Cells
The roles of ERK1/2 and MEK1/2 activation in the LDR-stimulating effect were examined using U0126, a specific inhibitor of MEK. As described previously, we observed the stimulation of ERK1/2 in the cells exposed to 50 mGy X-irradiation compared to the cells without radiation. The stimulating effect of ERK1/2 by LDR was significantly suppressed by the addition of U0126 in a dose-dependent manner (Figure 4A and B).
The PI3K/AKT pathway has also been reported to be involved in regulating the expression of MEK 10 ; therefore, we also utilized the specific inhibitor of PI3K, LY294002, to examine its effects on AKT phosphorylation. The results demonstrated that exposure to 50 mGy X-irradiation significantly increased AKT phosphorylation levels compared to those in nonirradiated cells; however, this stimulation was suppressed by LY294002 in a dose-dependent manner (Figure 4C and D).
The suppressive effects of the inhibitors on enhanced cell proliferation mediated by 50 mGy X-ray exposure are shown in Figure 4. Both 10 μmol/L U0126 and 10 μmol/L LY294002 diminished the stimulation of cell proliferation (Figure 4E), which suggests that LDR-stimulated cell proliferation is ERK1/2 and AKT activation dependent.
Discussion
Radiotherapy is very important for treating malignant tumors and is either used following surgery or alone for inoperable tumors. The efficacy of radiotherapy is limited by associated normal tissue toxicity as well as by tumor resistance. 11,12 Conversely, experimental studies with low-dose γ- and X-irradiation in various laboratory animals and cell lines have demonstrated the phenomenon of hormesis and adaptive response in normal cells and tissues but not in malignant tumors. 13,14
The experimental data obtained from the present study confirm that LDR induces cell proliferation in the normal human embryonic lung fibroblast 2BS cell line. This is consistent with the results from our previous work, which showed the stimulating effects of LDR on mouse hematopoietic progenitor cells 15 and rat mesenchymal stem cells. 7 Several other studies have also reported the observation of cell proliferation induced by LDR in normal cells including normal human diploid cells and Chinese hamster fibroblasts. 13,14,16 In our study, measurements of total cell numbers, CCK-8 assessment of proliferation, and cell cycle distribution all indicated that LDR enhanced 2BS cell proliferation. As seen in Figure 2, there was a significant increase in the proportion of 2BS cells in the S-phase between 2 and 6 hours after exposure to 50 mGy X-irradiation compared to unexposed controls, suggesting that LDR promoted transient cell proliferation. In the lung cancer cell line NCI-H446, however, LDR did not induce cell proliferation and even induced apoptotic cell death (Figure 2). Some investigators have noted the extreme sensitivity of cells to LDR, defined as hyperradiosensitivity. 17,18 This suggests that 2BS and NCI-H446 have different responses to LDR.
Previously, Khodarev et al reported differences in the dose-dependent and independent temporal patterns of gene response to ionizing radiation in normal versus tumor cells. 19 However, in that early study, the lowest dose of radiation utilized was 1.0 Gy. Therefore, whether there is also a difference in the responses of normal and tumor cells to LDR remains unclear. The present study indicated for the first time, as shown in Figure 3, that the MAPK/ERK pathway showed a significant difference between normal and tumors cells in its response to LDR at 20 to 75 mGy X-irradiation. Our results demonstrated that 2BS cells, but not NCI-H446 cells, showed significant increases in c-Raf/MAPK/MEK/ERK and PI3K/PDK-1/AKT pathway activation following LDR.
The MAPK/ERK activation has been previously observed by our group in rat mesenchymal stem cells. 7 Another study has shown that transient activation of Raf resulted in the activation of ERK following growth factor stimulation and cell cycle progression. 20 Furthermore, several studies have also demonstrated that inhibition of MAPK leads to cell death. 13,21 One study using normal human diploid cells demonstrated that the activation of ERK1/2 by 20 mGy X-irradiation resulted in a stimulation of cell proliferation, suggesting that ERK1/2 activation has cell survival effects. 13 However, whether the MAPK pathway activation only occurs in normal cells but not in tumor cells in response to LDR has not been directly compared under the same experimental conditions in previous studies. In the present study, we demonstrated that the activation of the ERK pathway was directly associated with 2BS cell proliferation, whereas in NCI-H446 cells, neither activation of ERK pathway nor stimulation of cell proliferation in response to LDR could be observed in the tested experimental condition. The coordinated activation of the c-Raf/MAPK/MEK/ERK network by LDR was further supported by the finding that inhibition of MEK with U0126 prevents the phosphorylation of ERK (Figure 4). Experimental evidence showed close interrelationships between Ras/Raf/MEK/ERK activation and cell cycle regulation. However, the inseparable links between P53, a crucial cell cycle regulator, and the MAPK/ERK and PI3K/AKT signaling pathways are complicated. 22,23
Another new finding of the present study is the enhanced phosphorylation levels of PDK-1 and AKT in 2BS cells exposed to LDR (50 mGy X-rays; Figure 5). The coordinated activation of the PI3K/PDK-1/AKT network by LDR is further supported by the outcome that blocking PI3K with LY294002 inhibited the phosphorylation of AKT. Kim et al have investigated the proliferation of CCD-18 Lu cells, another normal human lung fibroblast cell line, under LDR. 24 Their results indicated that the phenomenon was associated with the activation of Raf and AKT. It is known that inhibition of Raf and AKT leads to ionizing radiation-induced cytotoxicity. 25 Reportedly, the PI3K/AKT signaling pathway also plays an important role in cell survival and against cell killing, and this effect might be related to Raf and ERK function. In addition, we found that blocking PI3K/AKT activation with LY294002 eliminated LDR-induced ERK activation (Figure 4E). Therefore, the present study suggests a cooperative function between the MAPK/ERK and PI3K/AKT signaling pathways in the development of LDR-mediated cell proliferation. The detailed mechanism underlying such cooperation will be determined in future studies.
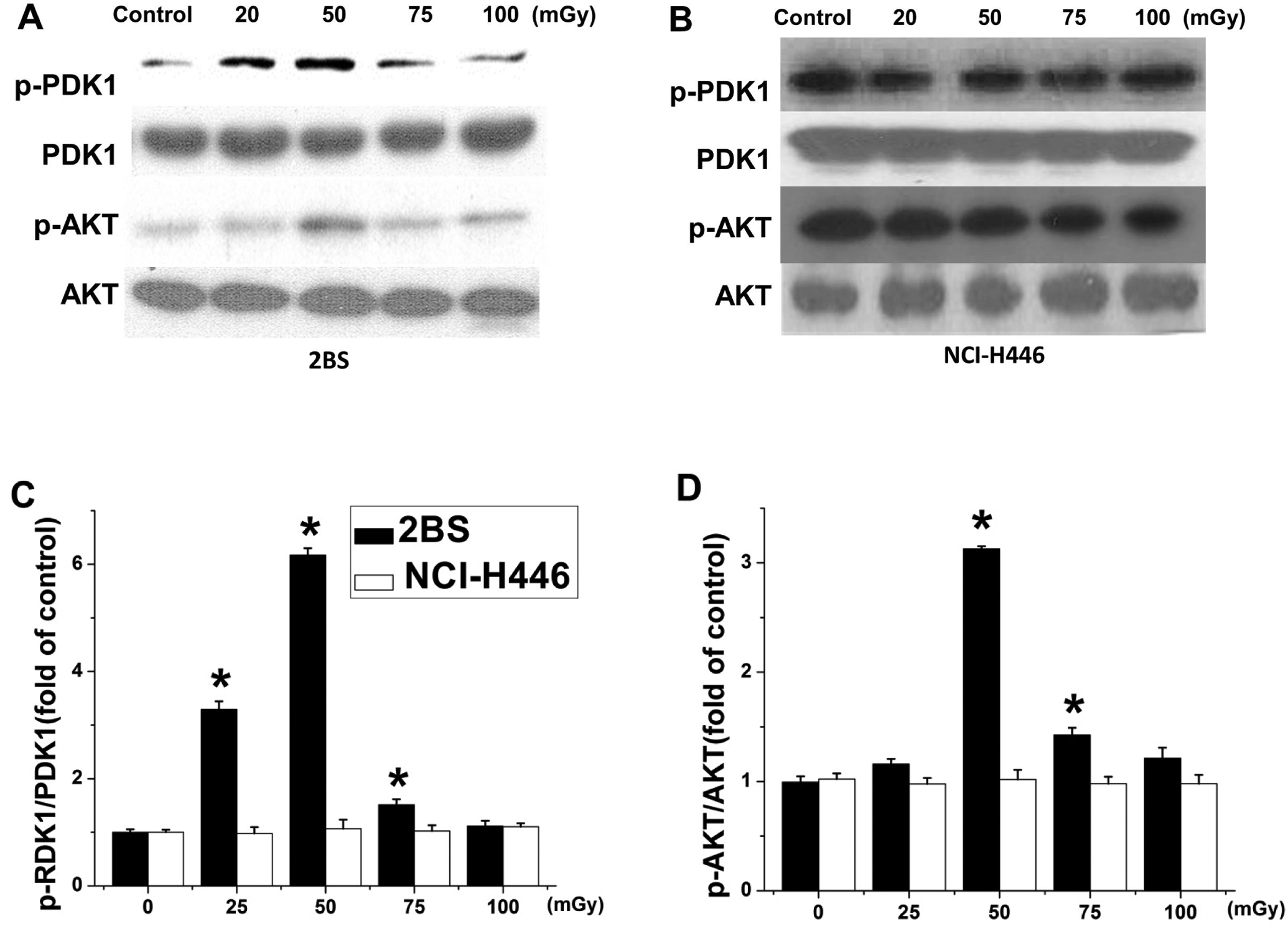
Low-dose ionizing radiation (LDR) induced phosphorylation of PDK-1 and AKT in 2BS but not NCI-H446 cells. Phosphorylation levels of PDK-1 and AKT were determined 6 hours after exposure to 20, 50, 75, or 100 mGy X-irradiation by Western blot analysis for both 2BS (A) and NCI-H446 (B) cell lines. The phosphorylated to total expression ratios of PDK-1 (C) and AKT (D) were quantitatively analyzed. Data are presented as the means ± standard deviation (SD) of 3 separate experiments with duplicate samples at a minimum. *P < .05 vs control.
The data for the expression of MAPK/ERK and PI3K/AKT pathway proteins showed no change in the NCI-H446 cell line after LDR, suggesting that LDR does not affect these signaling pathways in NCI-H446 cells. However, complicated relationships between hormesis, adaptive response, and hyperradiosensitivity might exist in these cells. Continued studies aimed at elucidating these connections at the cellular and molecular levels will be necessary to understand these phenomena more comprehensively.
Investigations during the last decade have shown that LDR can enhance the immune response. 26,27 Low-dose radiation activates several types of immune cells including dendritic cells, natural killer cells, T cells, and macrophages and leads to an increase in mast cell activity. 28,29 In a study from the United States that reviewed the incidence of lung cancer in high-impact radiation states where nuclear testing occurred, the results suggested that exposure to LDR might be protective against lung cancer rather than a cause thereof. 30 These advances in our understanding of LDR-induced anticancer effects suggest that the clinical application of LDR is a feasible alternative to other traditional therapeutic modalities for the treatment of malignant tumors.
In summary, this study demonstrated that a proliferative response can be induced by exposure to 50 mGy X-irradiation in 2BS cells. Furthermore, we showed that the activation of MAPK/ERK and PI3K/AKT signaling pathways was indispensable for LDR-induced cell proliferation in 2BS cells, as illustrated in Figure 6; however, in NCI-H446 tumor cells, no hormesis was observed but rather a hyperradiosensitivity response to LDR was seen in which the MAPK/ERK and PI3K/AKT pathways were not involved. Together, these results suggest that the possibility of the clinical application of LDR needs to be explored in future studies.
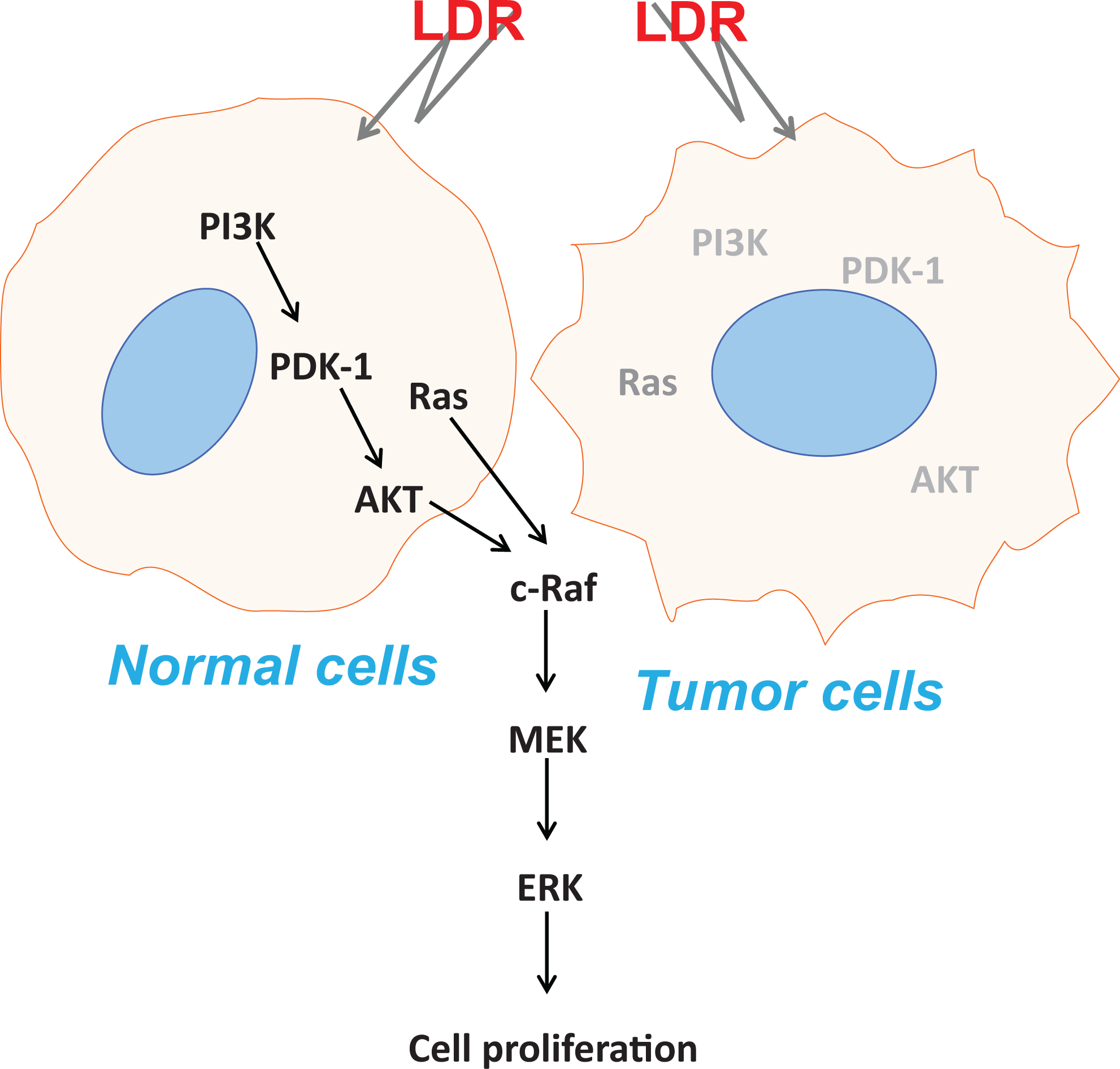
Illustration of working mechanisms by which low-dose ionizing radiation (LDR) might induce proliferation in normal but not tumor cells. Here, it is assumed that the status of whether LDR can activate Mitogen-activated protein kinases (MAPK)/extracellular signal-regulated kinase (ERK) and PI3K/AKT signaling pathways determines the induction of cell proliferation.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by grants from the National Science Foundation of China (81302379, X.L.; 81302380, D.Y.; 31300695, Q.L.), the Natural Science Foundation of Tianjin (13JCYBJC23500 and 13JCQNJC11600, Q.L.), and the Ministry of Education Key Project of Science and Technology (311015, J.C.).