
Abstract
Background
Microglial-induced inflammation plays a crucial role in the pathophysiological process of nervous system injury, however, still lacks effective therapeutic agents. Previously, we discovered that the inhibition of histone deacetylase 3 (HDAC3) exerts anti-inflammatory effects after traumatic spinal cord injury (SCI), whereas little is known about its underlying mechanism. Therefore, the present study aimed to explore the effects and potential mechanisms of HDAC3 on neuroinflammation and microglial function.
Methods
Rats were randomized into 4 groups: sham group, SCI group, SCI + vehicle group, and SCI + RGF966 group. To examine the effect of HDAC3 on neurological deficit after SCI, we gathered data using the Basso Beattie Bresnahan locomotion scale, the inclined plane test, the blood-spinal cord barrier, junction protein expression, and Nissl staining. We also evaluated microglial activation and inflammatory factor levels. Immunofluorescence analysis, immunohistochemical analysis, western blotting, and quantitative real-time polymerase chain reaction were performed to examine the regulation of the Sirtuin 1 (SIRT1)/nuclear factor-erythroid 2-related factor 2 (Nrf2) pathway.
Results
The results showed that HDAC3 inhibition significantly ameliorated Basso–Beattie–Bresnahan (BBB) permeability, brain edema, and improved neurological functions and junction protein levels. Additionally, HDAC3 inhibition significantly inhibited microglial activation, thereby reducing the levels of SCI-induced pro-inflammatory factors. Moreover, HDAC3 inhibition dramatically enhanced the expression of SIRT1 and increased both Nrf2 nuclear accumulation and transcriptional activity, thereby increasing downstream heme oxygenase-1 and NAD(P)H quinone oxidoreductase 1 expression.
Conclusions
The results of this study suggest that HDAC3 inhibition protects the spinal cord from injury following SCI by inhibiting SCI-induced microglial activation and the subsequent inflammatory response via SIRT1/Nrf2 signaling pathway, highlighting HDAC3 as a potential therapeutic target for the treatment of SCI.
Introduction
Traumatic spinal cord injury (SCI) is a complicated pathophysiological procedure involving the central nervous system (CNS), which causes the loss of nerve function, including those implicated in movement, sensation, and features a high mortality and disability rate.1,2 Unpredictable primary mechanical injury, which mainly damages the spinal cord directly, is a major factor determining the outcome of SCI. Accumulating studies have indicated that secondary nonmechanical injury caused by pathological processes; including oxidative stress, edema, excitotoxicity, and inflammation; plays a crucial role after the primary insult.3 -5 Increasingly, research has targeted microglial activation and the subsequent inflammatory reaction, which increase capillary permeability, disrupt the blood-spinal cord barrier, ultimately impede axonal regeneration and neural functional recovery.6 -8 Previous studies on SCI have revealed that the inhibition of microglial activation and the subsequent neuroinflammatory response could restrict the extent of tissue injury and thus protect against disability. 9
Histone deacetylase 3 (HDAC3) is a class I HDAC that is highly expressed in the hippocampus, cortex, and cerebellum, participates in the occurrence and development of tumorigenesis, inflammation, and cardiovascular and nervous system diseases.10,11 HDAC3 inhibition generates chromatin protein hyperacetylation and alterations in gene expression, which are deemed to be potential therapeutic targets for a variety of diseases.12 -14 Recently, experimental studies have shown that HDAC3 inhibitors exert neuroprotective effects against inflammatory cytokines released in various neurological diseases. 15 Additionally, our group previously reported that HDAC3 inhibition protects neurons from inflammatory injuries following SCI. 16 However, few studies addressed the exact function of HDAC3 and its underlying mechanisms following SCI.
Sirtuin 1 (SIRT1) is a nicotinamide adenine dinucleotide (NAD)-dependent histone deacetylase (HDAC) and an important regulatory factor in oxidative metabolism and inflammatory signaling pathways. 17 Numerous studies have demonstrated that SIRT1 is involved in the regulation of different biological processes, such as cell proliferation, neuronal function, mitochondrial biogenesis, and inflammation.18 -20 SIRT1 has also been reported to exert protective effects in many CNS diseases, including Alzheimer’s disease, subarachnoid hemorrhage, and traumatic brain injury (TBI).21 -23 Our previous work found that the expression of SIRT1 was significantly increased after TBI, indicating a possible role of SIRT1 after TBI. Furthermore, SIRT1 could protect neuronal survival against nuclear factor kappa B-induced inflammatory pathways associated with neuronal apoptosis. 24 These emerging experimental findings suggest that SIRT1 plays an important role in the pathogenesis of SCI. Nevertheless, its molecular mechanisms remain elusive.
Nuclear factor-erythroid 2-related factor 2 (Nrf2) is a transcription factor that modulates cellular redox homeostasis, and coordinately regulates the transcription of many antioxidants and phase 2 detoxification enzymes, such as heme oxygenase-1 (HO-1) and NAD(P)H quinone oxidoreductase 1 (NQO1).25,26 Studies have shown that Nrf2 is indispensable for bodily defense against oxidants and inflammatory insults after SCI. 27 Thus, Nrf2 has been proposed to be a promising target for attenuating inflammation. Importantly, Nrf2 expression levels have been correlated to HDAC enzymatic activity and the levels of histone acetylation. 28 These emerging experimental findings imply that HDAC3 is likely to play an important role in diabetic pathophysiology and its complications.
Considering that the HDAC3 is a vital regulatory factor of SIRT1 and Nrf2 expression, and the crucial role of microglial-induced inflammation following SCI, we speculated that HDAC3 may exert its neuroprotective effect by modulating microglial activation via the SIRT1/Nrf2 pathway. To address this hypothesis, we explored the curative effect of HDAC3 against secondary injury of SCI and the underlying molecular mechanisms.
Materials and Methods
Animal Preparation
All experimental protocols were approved by the Ethics Committee at Fujian Medical University (Fujian, China). Adult male Sprague–Dawley rats (weighing 220-250 g) were purchased from the Fujian Medical University Laboratory Animal Center. The rats had ad libitum access to food with a 12-hour light/dark cycle at 22°C throughout the experiment. All animal procedures were approved by the Institutional Animal Care and Use Committee at the Fujian Medical University. All experimental procedures complied with recommendations from the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and ease animal suffering as much as possible.
Model of SCI and Drug Delivery
The traumatic SCI model used in this study was established in accordance with previous literature. 29 No sample size calculation was performed. Briefly, rats were anesthetized with the inhalation of isoflurane (2% in oxygen gas, 300 ml/minutes) after being placed in a stereotaxic frame. After disinfection, approximately 2.0 cm back midline incision was made along vertebrae T8 to T11 to expose the vertebral laminas. The thoracolumbar fascia and paravertebral muscles were peeled along the spinous processes. Laminectomy was performed along vertebrae T9 to T10, maintaining the integrity of the dura mater in the exposing procession. The T10 level of the spinal cord (1.5 mm lateral to the midline) was located and subjected to weight-drop impact to induce contusion injury (10 g × 25 mm) by a 10-g metal rod. The muscles and skin were sutured in layers, and the animals were placed in quondam cages. The sham group only sustained laminectomy without injury. There was no sedative or analgesic drugs which maybe cause postponed wakening were given after the operation according to large amounts of previous studies (Chen et al 16 , Lv et al 6 , and Parra-Villamar et al 5 ) Postoperative manual bladder emptying was performed twice daily until the animals could urinate by themselves.
The concentration and dose selection of RGFP966 (an HDAC-inhibitor with selectivity for HDAC3) was determined according to a previous study. Briefly, 1% RGFP966 [dissolved in dimethyl sulfoxide (DMSO, Sigma–Aldrich, St Louis, MO, USA, D8418, <10% final volume) diluted with 30% hydroxypropyl β-cyclodextrin] was injected intraperitoneally 30 minutes after SCI. RGFP966 (or a vehicle) was administered at the dosage of 10 mg/kg/day by intraperitoneal injection for 3 consecutive days.
Experiment Design
Part 1: A total of 24 rats (26 rats were used, 2 rats died) were randomly assigned to 4 groups: the sham group, SCI group, SCI + vehicle group, or SCI + RGFP966 group (n = 6 per group). The locomotor recovery was evaluated at 1, 3, 7, and 14 days after SCI. Post assessments included a behavioral assessment.
Part 2: A total of 48 rats (55 rats were used, 7 rats died) were randomly assigned to 4 groups: the sham group, SCI group, SCI + vehicle group, or SCI + RGFP966 group (n = 12 per group). All the rats were sacrificed 3 days after SCI according to the results of the first experiment. Post assessments included a behavioral assessment, spinal cord water content measurements, and blood-brain barrier permeability determination.
Part 3: A total of 72 rats (81 rats were used, 9 rats died) were randomly allocated into 4 groups: the sham group, SCI group, SCI + vehicle group, or SCI + RGFP966 group (n = 18 each). Their tissues were then processed for subsequent assessments, including immunofluorescence analysis, western blot analysis, Nissl staining, quantitative real-time polymerase chain reaction (qRT-PCR), enzyme-linked immunosorbent assay (ELISA), and immunohistochemical staining.
Nuclear, Cytoplasmic, and Total Protein Extraction
After anesthesia with isoflurane, animals were perfused intracardially with saline (4°C). Tissues were isolated and processed using the homologous protein extraction kit. Spinal cord tissues were immersed in 20 mmol/l Tris (pH 7.6) and centrifuged for 15 minutes at 14 000g at 4°C. The resulting supernatant was collected and stored at −80°C. To extract the cytosolic proteins, the tissue samples were lyzed in hypotonic lysis buffer A (20 M HEPES (pH 7.4), 2 mM EGTA, and 2 mM MgCl2). The mixture was then centrifuged for 10 minutes at 5000g and 4°C after 30 μl of 10% NonidetP-40 solution was added. The resulting liquid supernatant was then collected and stored at −80°C. To extract the nuclear proteins, the tissue samples were lyzed using buffer B (20 mM Tris/HCl, pH 7.6, 100 mM NaCl, 20 mM KCl, 1.5 mM MgCl2, 0.5% Nonidet P-40, and protease inhibitors). After the mixture was vortexed for 30 seconds and spun by centrifugation for 10 minutrd at 5000g and 4°C, the cytosolic fraction extracts were collected and stored at −80, 4°C. Protein concentrations were measured using the BCA method (Beyotime Biotec, Jiangsu, China).
Neurological Behavior
The behavioral function of rats was tested at 1, 3, 7, and 14 days using the BBB locomotion rating scale following the previous studies. The score was determined by the athletic ability of rats, including hind limb joint movements, motor function, and coordination of forelimbs. No score was given for failing on tasks; a score of 0 was given for complete paralysis; a score of 21 was given for normal locomotion. Behavioral assessments were performed by 2 observers who had no prior knowledge of the group information, and the final scores were given by consensus.
Spinal Cord Water Content
Rats were anesthetized as described above. The brain cerebrum and cerebellum were removed, and the spinal cord segments containing the lesion were separated and weighed immediately (wet weight, ww). Each part was weighed and parched at 80°C for 72 hours, before being re-weighed (dry weight, dw). The percentage of water content was calculated as [(ww − dw)/ww] × 100%.
Measurement of Blood-Spinal Cord Barrier (BSCB) Permeability
The permeability of the BSCB was evaluated by measuring the diffuse of Evans blue staining (Sigma–Aldrich). The Evans blue dye (2% in saline; 4 ml/kg) was transcardially perfused with atrium dextrum with phosphate-buffered saline (PBS), followed by additional PBS containing 4% paraformaldehyde. Each tissue sample was immediately weighed and homogenized in 1 mL of 50% trichloroacetic acid solution, then centrifuged. The absorption of the supernatant was measured by a spectrophotometer (UV-1800 ENG 240V; Shimadzu Corporation, Japan) at a wavelength of 620 nm. The quantity of Evans blue dye was calculated using a standard curve and expressed as micrograms of Evans blue dye per gram of brain tissue.
Nissl Staining
The paraffin-embedded sections of spinal cord specimens near the lesion epicenter cut (4 μm thick) were stained with Nissl staining solution (Boster Biotech, Wuhan, China) and then washed with double-distilled water after dehydration and fixed with Permount TM mounting medium. Staining was visualized and captured under a light microscope. The neurons were observed for their cellular morphology and counted using the ImageJ software. Normal neurons had large and full soma with a rich cytoplasm and 1 to 2 round, large nuclei. In contrast, apoptotic cells had shrunken neuronal cell bodies and contained dark cytoplasm vacuoles and hyperchromatic nuclei. The examination was performed by 2 independent observers who were blinded to the experimental groups. To quantify the amount of Nissl staining, 5 high-power areas (at ×400 magnification) were randomly selected, and the mean number of intact neurons in each view was regarded as the data for each sample.
TUNEL Analysis
The proportion of apoptotic cells was detected by a terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) detection kit (Roche, Indianapolis, IN, USA) according to the manufacturer’s instructions. Each section from the lesioned areas was incubated overnight with primary antibody NeuN (1:100; Boster Biotech) at 4°C. The sections were then washed three times with PBS for 45 minutes and blended with TUNEL reaction solvent for 1 hour at 37°C. The slides were then washed three times with PBS and counterstained by 4′,6-diamidino-2-phenylindole (DAPI). Five randomly selected areas were subsequently selected to identify, count, and analyze the TUNEL-positive neurons under a ZEISS HB050 fluorescence microscope. The final average percentage of the apoptotic index was regarded as the data for each sample.
Immunofluorescence Analysis
Immunofluorescence staining was performed as reported in a previous study. 30 Briefly, spinal cord specimens were fixed in 4% paraformaldehyde for 24 hours, permeabilized by 0.3% Triton X-100, blocked by 1% bovine serum albumin, embedded in paraffin, and cut to 5 μm. The sections were then blocked with buffer for 2 hours and subsequently incubated overnight at 4°C with primary antibodies. After washing the sections three times with PBS for 45 minutes, they were incubated with secondary antibodies (Alexa Fluor 488 and Alexa Fluor 594, 1:200) for 2 hours at room temperature. The slides were then counterstained with DAPI for 2 minutes after washing with PBS again three times for 45 minutes. The slides were then covered by a coverslip with an anti-fade mounting medium. Fluorescence microscopy imaging was performed using an inverted microscope system (Leica, Wetzlar, Germany) at ×400 magnification and analyzed using the Image Pro Plus 6.0 software (Media Cybernetics, Rockville, MD, USA).
Quantitative Real-Time Polymerase Chain Reaction
The total RNA obtained from the spinal cord tissues was extracted using Trizol (Invitrogen, USA) according to the manufacturer’s instructions. cDNA was subsequently reverse transcribed from the mRNA using the Prime Script RT reagent kit (Vazyme, Nanjing). qRT-PCR was performed in an Mx3000P PCR system (Stratagene, San Diego, CA, USA) in a final reaction volume of 25 μl containing 200 ng cDNA, 1.25 μl each of the 2 primers (stock concentration, 10 µM), and 12.5 μl of the SYBR Green RT-PCR master mix. The thermal cycling parameters were as follows: pre-denaturation at 95°C for 15 minutes, followed by 40 cycles of denaturation at 94°C for 15 seconds, annealing at 55°C for 30 seconds, and extension at 70°C for 30 seconds. The primers were as follows: NQO1 (F: 5′-CAT TCT GAA AGG CTG GTT TGA-3′; R: 5′-CTA GCT TTG ATC TGG TTG TCAG-3′), HO-1 (F: 5′-ATC GTG CTC GCA TGA ACA CT-3′; R: 5′-CCA ACA CTG CAT TTA CAT GGC-3′), and β-actin (F: 5′-CGT GAA AAG ATG ACC CAG ATCA-3′; R: 5′-CAC AGC CTG GAT GGC TAC GTA-3′). The results were analyzed in triplicate using the 2−ΔΔCt method, while 18S RNA was used for normalization.
Enzyme-linked immunosorbent assay
The spinal cord specimens were homogenized in lysis buffer containing proteinase inhibitor. The resulting homogenates were centrifuged at 12 000g for 20 minutes at 4°C. The protein concentrations were then determined using the BCA method (Beyotime Biotec, Jiangsu, China). The levels of TNF-α, IL-1β, and IL-6 in the spinal cords were quantified using specific ELISA kits (Biocalvin Company, Suzhou, China) according to the manufacturer’s instructions.
The Multiplex Microplate Reader (Molecular Devices, SpectraMax M5) was used to detect the absorbance values of the ELISAs. The OD values were determined and expressed as pg/mg protein.
Western Blot Analysis
Equal protein amounts were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. The membranes were blocked in 5% nonfat milk for 2 hours and then incubated with primary antibodies against Nrf2 (1:1000, Abcam, Cambridge, MA, USA), Bcl-2, SIRT1, Iba-1, GFAP (1:200, Santa Cruz Biotechnology, CA, USA), TNF-α, IL-6, IL-1β (1:400, Cell Signaling Technology, Danvers, MA, USA), cleaved caspase-3 (1:1000, Cell Signaling Technology, Danvers, MA, USA), HO-1 (1:200, Santa Cruz Biotechnology), Bax, NQO1, Histone H3 (1:1000, Abcam, Cambridge, MA, USA), or β-actin (1:5000, Bioworld Technology, St. Louis Park, MN, USA) antibodies in blocking buffer. The membranes were washed three times with TBST for 15 m and then incubated in secondary antibodies conjugated with horseradish peroxidase-conjugated IgG (1:1000, Bioworld Technology, St. Louis Park, MN, USA) for 2 hours at room temperature. The immunoblots were visualized using the Millipore ECL Western Blotting Detection System (Millipore, Billerica, MA, USA). Band densities were quantified using the UN-Scan-It 6.1 software (Silk Scientific Inc., Orem, UT, USA)
Immunohistochemical Staining
The tissue sections (4 mm thick) were incubated overnight with a primary antibody against Nrf2 (1:100, Abcam, Cambridge, MA, USA). The sections were then incubated with horseradish peroxidase-conjugated IgG (1:400, Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 60 minutes at room temperature after washing three times (15 minutes each) in PBS. After washing for half an hour with PBS, diaminobenzidine was used as a chromogen for the immunolabeled protein and counterstaining was performed using hematoxylin. The negative control was performed following the same steps without the primary antibody. Microscopy of the tissue sections was conducted by 2 pathologists blinded to the experimental process. The evaluation of sections was undertaken by assessing the intensity of staining (5 grades): 0—no detectable positive cells; 1—very low density of positive cells; 2—moderate density of positive cells; 3—higher density of positive cells; and 4—highest density of positive cells.
Statistical Analysis
All data were presented as the mean ± standard error of the mean (SEM). The measurements were subjected to a 1-way analysis of variance followed by Tukey’s post hoc test. Differences between 2 experimental groups were determined using the Student’s t-test. P < .05 was considered to indicate statistical significance.
Results
HDAC3 Inhibition Reduced Spinal Cord Edema and Neurological Function
The data in Figure 1A are analyzed appropriately with repeated-measures ANOVA. The BBB scores were assessed to evaluate the effect of locomotion recovery after SCI. The similar BBB scores of the sham groups were obtained at all time points. However, spinal cord insults caused the lowest score immediately compared with the sham group after surgery. The BBB scores of rats in these 3 groups gradually increased over time, with a significant improvement observed in the SCI + RGFP966 group 7 days following SCI (Figure 1A). The results of the behavioral assessment demonstrated that HDAC3 inhibition exerted a protective effect in the lesioned spinal cord 7 days after the SCI. Accordingly, subsequent experiments would be performed 7 days after the SCI. Spinal cord edema is an independent risk factor for negative outcomes following SCI. As shown in Figure 1B, the water content of the sham group was relatively low. Meanwhile, the tissue surrounding the lesion was significantly increased after SCI. The suppression of HDAC3 by RGFP966 crippled the water content compared with the SCI + vehicle group.
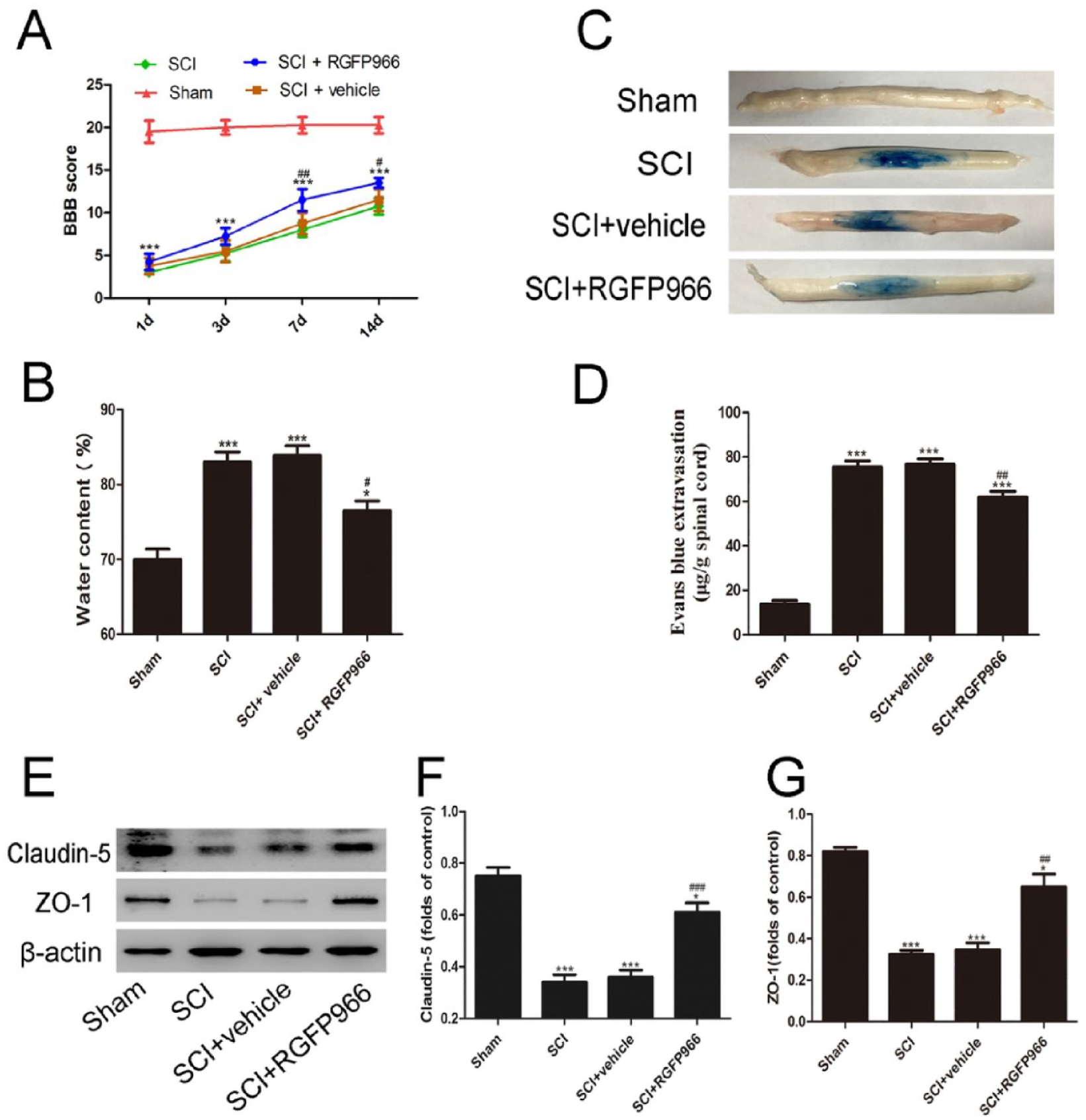
Inhibition of HDAC3 played protective effects against SCI. (A, B) Effects of RGFP966 on BBB scores and water content after SCI. (C, D) Representative photos of the Evans blue dye extravasation in the experimental groups 7 days after the SCI. (E-G) Representative Western blot images and analysis quantification of junction proteins ZO-1 and Claudin-5. Data are expressed as the mean ± SEM.
HDAC3 Inhibition Reduced SCI-Induced BSCB Permeability and Junction Protein Expression
Extravasation of the Evans blue was detected to investigate blood-spinal cord barrier (BSCB) permeability following SCI. Compared with the sham group, the SCI and SCI + vehicle group exhibited substantially more Evans blue dye extravasation 3 days after SCI. Moreover, HDAC3 inhibition by RGFP966 significantly decreased the extravasation of Evans blue dye compared to the vehicle group (Figure 1C,D). Tight junction proteins play important roles in maintaining BBB integrity, and these were detected by western blot. The results showed that the levels of ZO-1 and Claudin-5 were significantly decreased after SCI, while HDAC3 inhibition significantly rescued their expression (Figure 1E-G).
HDAC3 Inhibition Promoted Neuronal Survival After SCI
Nissl staining and TUNEL analysis were used to identify the apoptotic neurons 7 days after SCI. The sham group showed integrated and clear neurons. Compared with the sham group, the percentage of shrinking cells was higher in the SCI group and SCI + vehicle group. The apoptotic fraction declined significantly after RGFP966 administration (Figure 2). The expression of apoptosis-associated proteins was measured by western blot and immunofluorescence. The results revealed that SCI caused the upregulation of cleaved caspase-3 and the pro-apoptotic protein Bax. Compared with the SCI + vehicle group, HDAC3 inhibition by RGFP966 significantly diminished the levels of cleaved caspase-3 and Bax, while enhancing the level of the anti-apoptotic factor Bcl-2. In addition, compared with the SCI + vehicle group, HDAC3 inhibition significantly elevated the number of caspase-3-positive neurons (Figure 3).
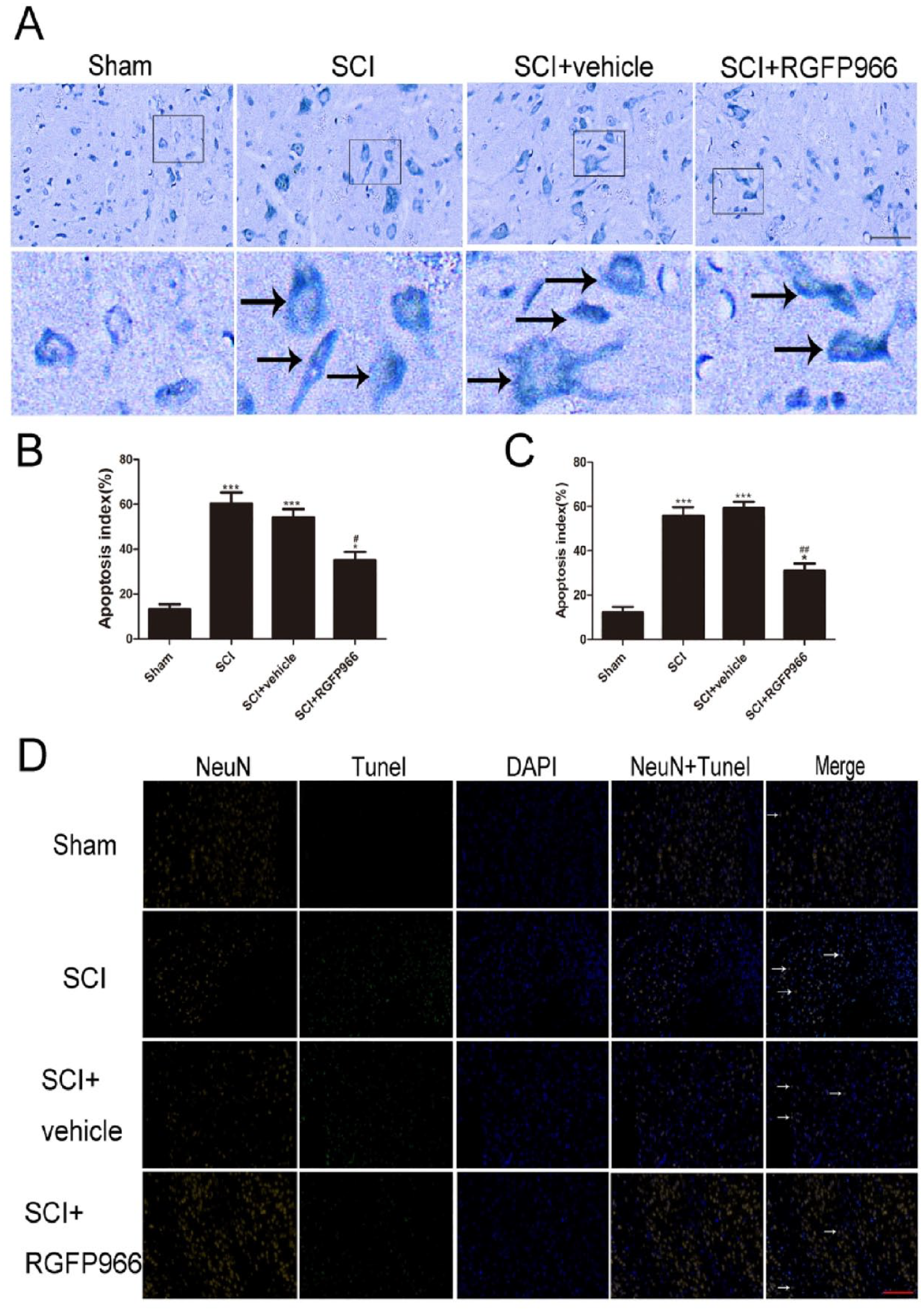
Inhibition of HDAC3 mitigated SCI-induced neuronal apoptosis in lesion spinal cord after SCI. (A, B) Representative photomicrographs and quantitative analysis of the Nissl staining after SCI. Enlarged images of boxed areas are shown below. Black arrows indicate the apoptotic neurons. (C, D) Representative photomicrographs and quantification of TUNEL staining. White arrows indicate apoptotic neurons. Data are expressed as the mean ± SEM.
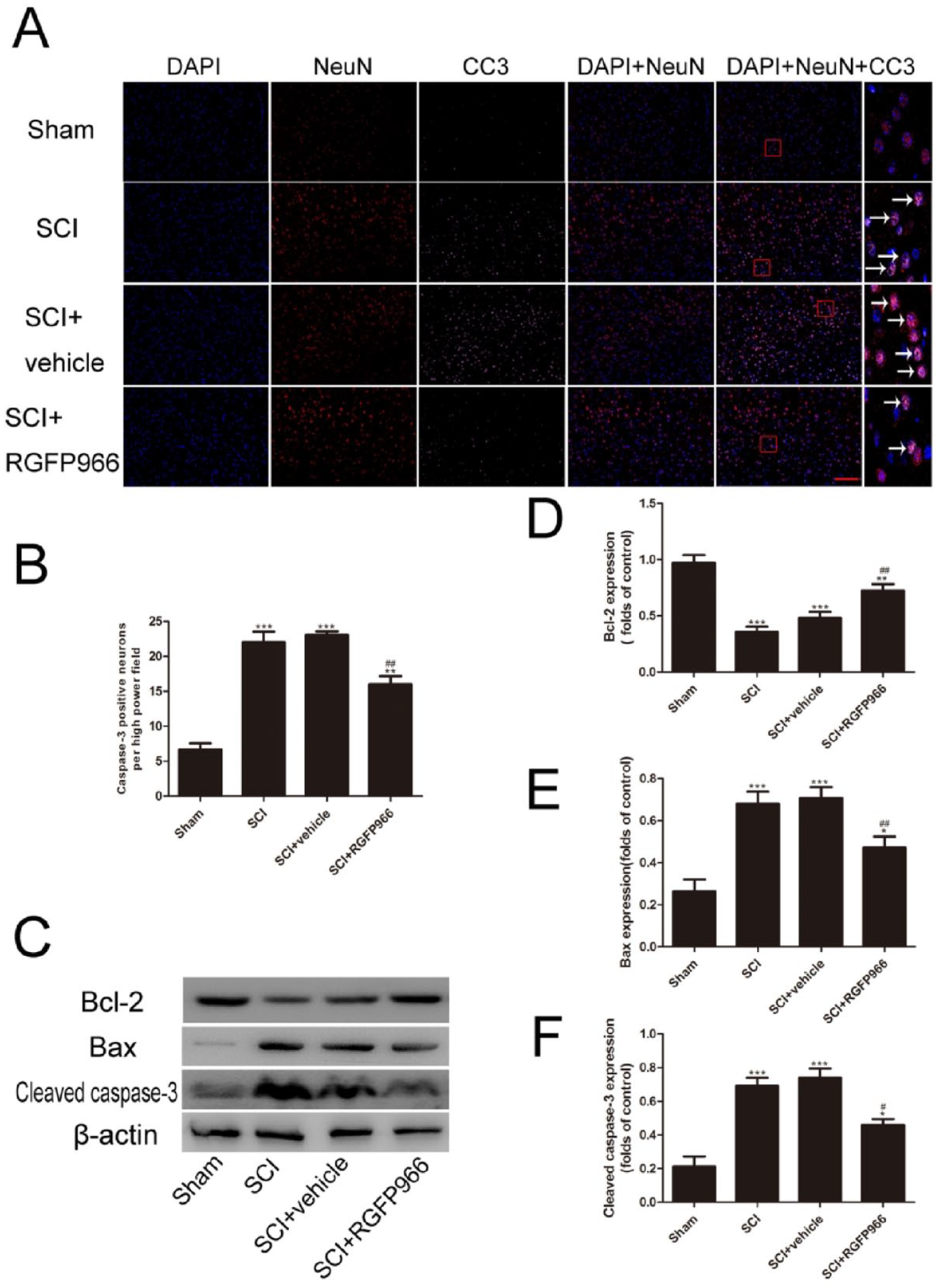
Inhibition of HDAC3 attenuated excessive inflammation after SCI. (A, B) Representative photomicrographs of immunofluorescent staining and quantitative analysis of caspase-3. White arrows point to positive neurons. (C, D) Representative Western blot gel images and quantification of Western blot analysis of Bax, Bcl-2, and cleaved caspase-3 expression in lesioned spinal cord. Data are expressed as the mean ± SEM.
HDAC3 Inhibition Reduced Microglial Activation and the Microglia-Mediated Inflammatory Response
Considering the effects of microglial activation and the subsequent inflammatory response following SCI, the effect of HDAC3 inhibition on microglial activation and expression of inflammatory factors were investigated. Both immunofluorescence analysis and western blot analysis showed that activated microglial cells (Iba-1+) increased after SCI. Interestingly, HDAC3 inhibition significantly inhibited microglial activation (Figure 4). The expression of microglia-mediated inflammatory factors (TNF-α, IL-1β, and IL-6) was measured using western blot analysis and ELISA. Compared with the sham group, the SCI group, and SCI + vehicle group had significantly higher expression levels of inflammatory factors, while HDAC3 inhibition ameliorated the SCI-induced enhancement of these factors (Figure 5).
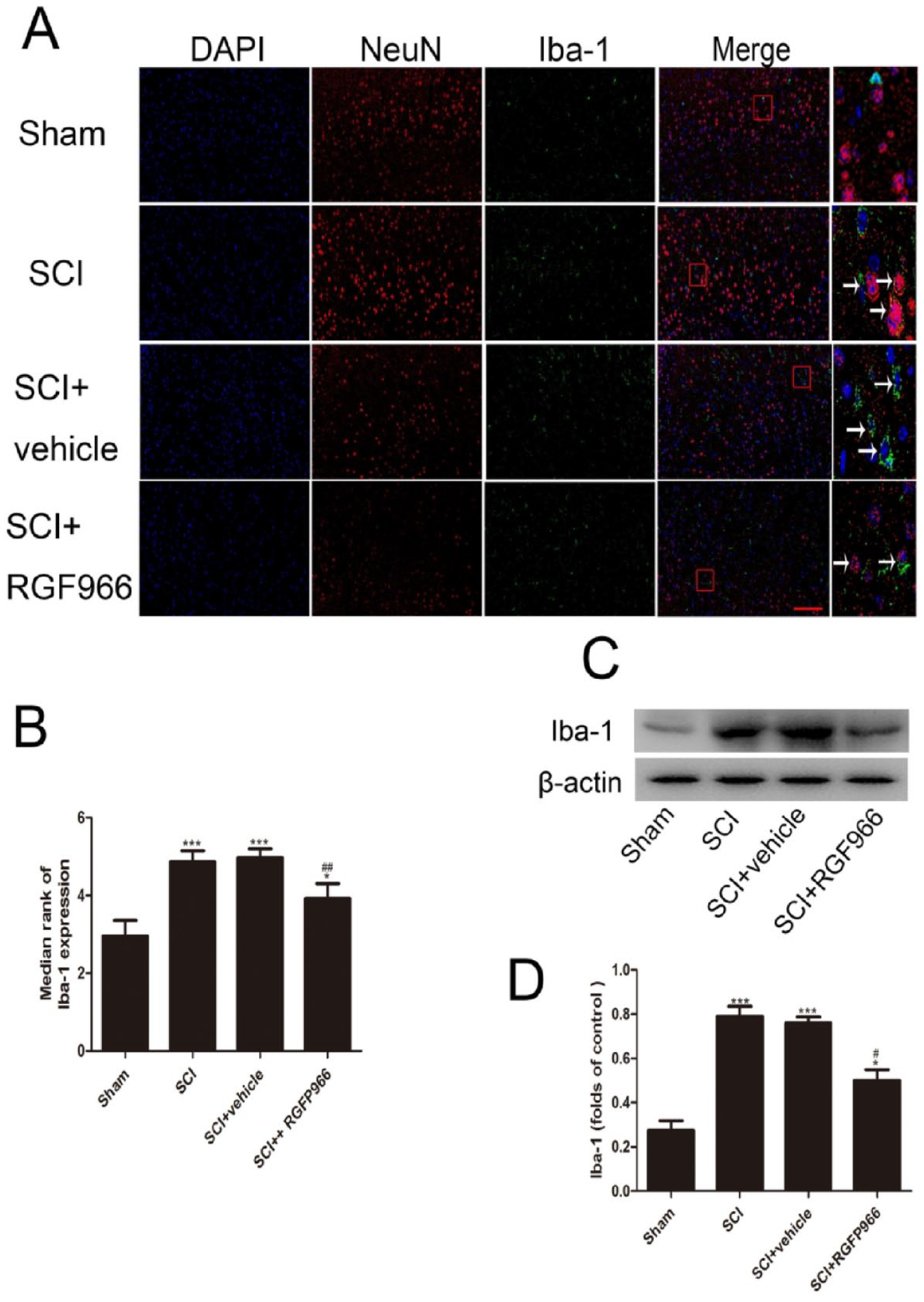
Inhibition of HDAC3 inhibited microglial activation in lesion spinal cord after SCI. (A, B) Representative photomicrographs and quantification of Iba-1 immunofluorescent staining. (C, D) Representative Western blot gel images and quantification of Western blot analysis of Iba-1. Data are expressed as the mean ± SEM.
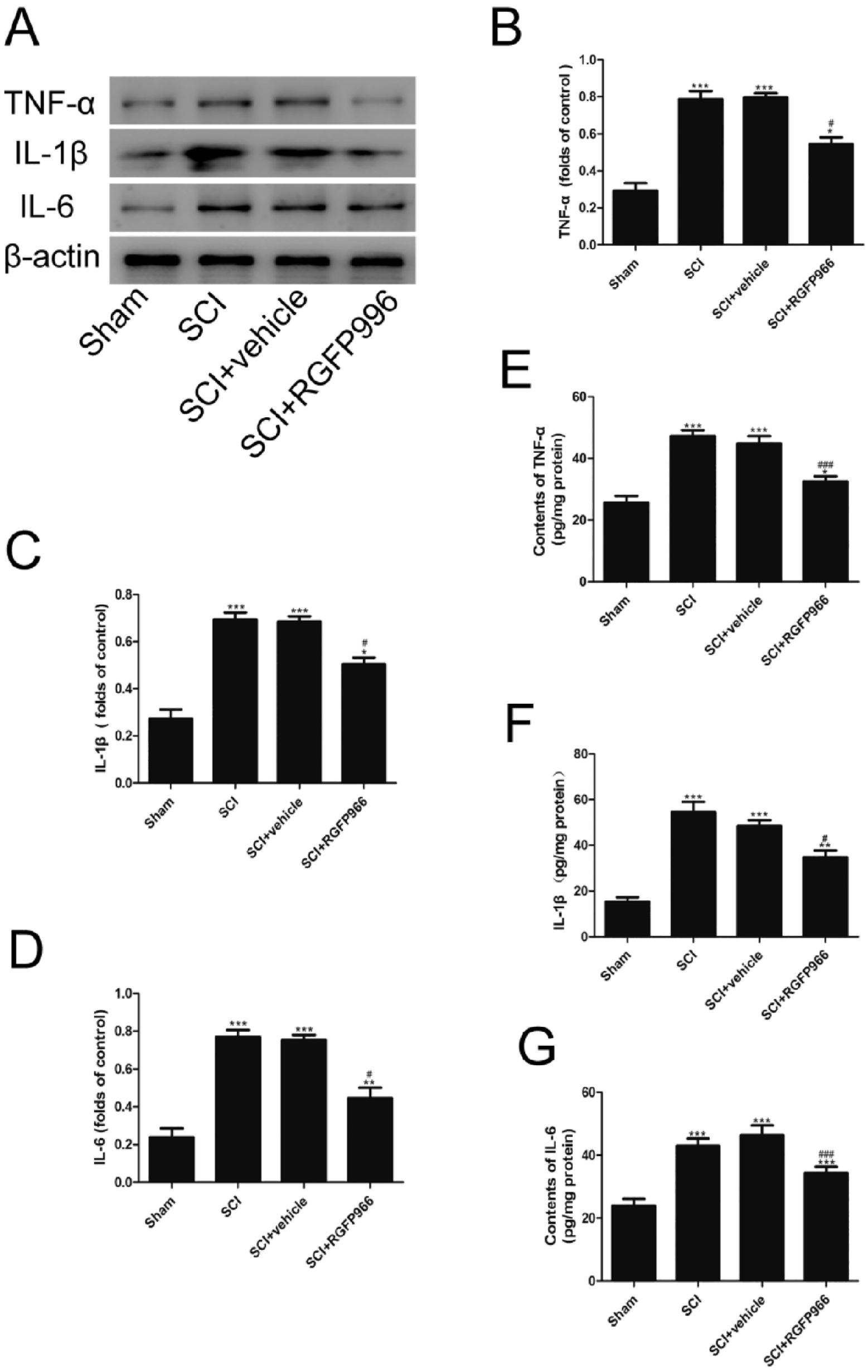
Inhibition of HDAC3 inhibits subsequent inflammatory response in lesion spinal cord after SCI. (A-D) Representative Western blot gel images and quantification of Western blot analysis of TNF-α, IL-1β, and IL-6. (E-G) TNF-α, IL-1β, and IL-6 protein expression were analyzed by ELISA.
HDAC3 Inhibition Facilitated SIRT1 Expression in Lesioned Spinal Cords
To investigate the mechanism of HDAC3 following SCI, the effects of HDAC3 inhibition by RGFP966 on SIRT1 expression were evaluated. The results of immunohistochemical staining showed that SIRT1 was expressed weakly in the sham group, but increased in the lesioned spinal cords after SCI (Figure 6A,B). Treatment with RGFP966 enhanced SIRT1 protein expression, which was reduced after SCI (P < .05). The results of the western blot analysis showed that SCI accelerated the expression and nuclear translocation of SIRT1. In addition, HDAC3 inhibition promoted SIRT1 expression compared with the SCI and SCI + vehicle groups. Compared with the SCI + vehicle group, HDAC3 inhibition promoted the nuclear translocation of SIRT1 (Figure 6C-F).
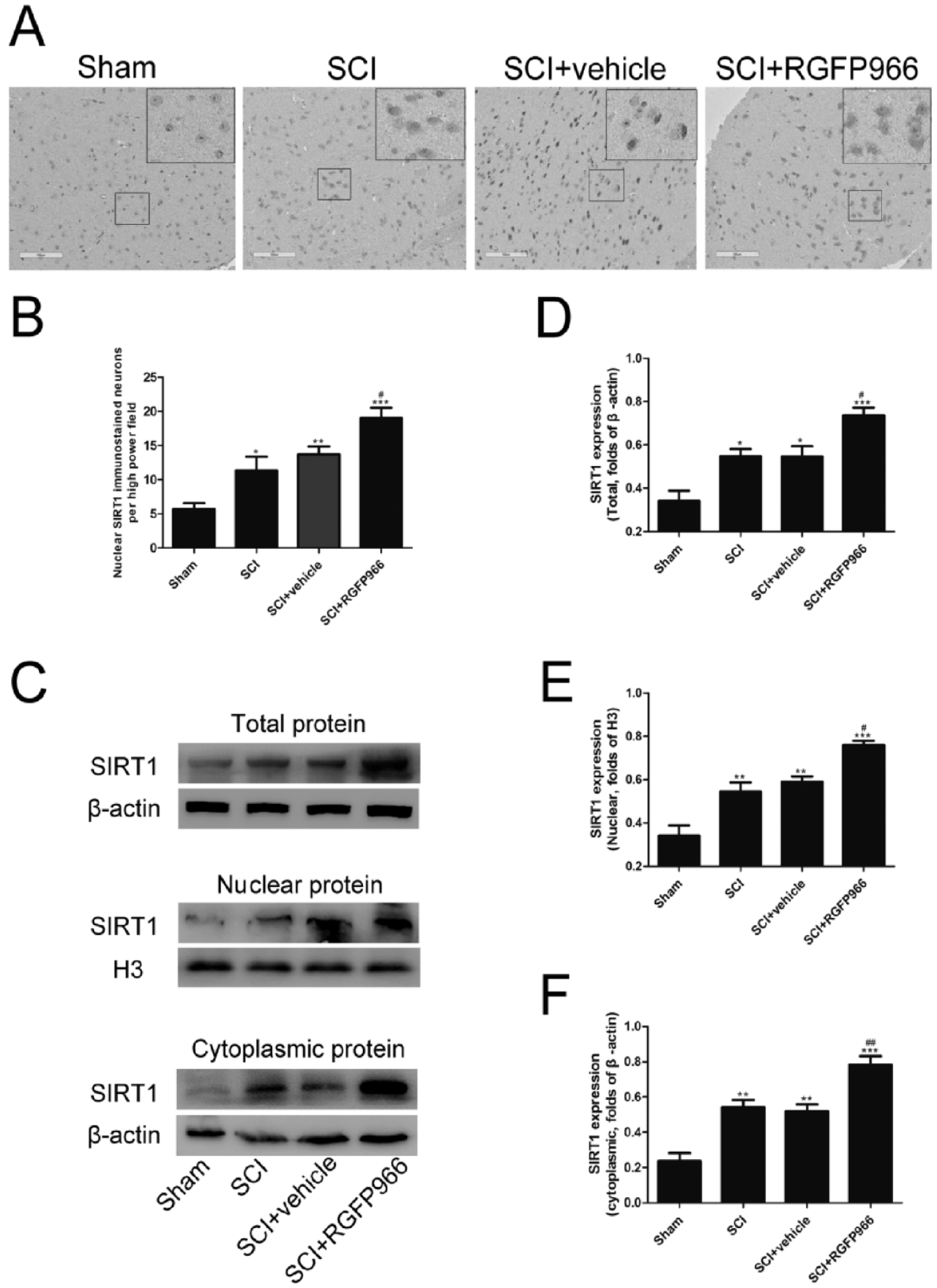
Inhibition of HDAC3 elevated SIRT1 expression and nuclear translocation in the lesion spinal cord after SCI. (A, B) Representative photomicrographs of immunohistochemical staining and quantitative analysis of SIRT1. (C-F) Representative Western blot gel images and quantification of Western blot analysis of total, nuclear, and cytoplasmic SIRT1 protein. Data are expressed as the mean ± SEM.
HDAC3 Inhibition Promoted the Nrf2 Pathway in Lesioned Spinal Cords After SCI
Changes in Nrf2 signaling following SCI were investigated in order to further obtain whether HDAC3 inhibition induced the activation of the Nrf2 pathway after SCI. Immunofluorescence analysis and western blot analysis both showed that Nrf2 was weakly expressed in the sham group and increased significantly after SCI (Figure 7). Compared with the SCI + vehicle group, significant augmentation of Nrf2 expression was observed in the SCI + RGFP966 group. The RGFP966 treatment promoted the nuclear translocation and expression of Nrf2. The levels of NQO-1 and HO-1 were also detected by qRT-PCR and western blot analysis. NQO-1 and HO-1 expression aggrandized significantly after SCI (Figure 8), and HDAC3 inhibition reduced their protein levels compared with the SCI + vehicle group. In addition, the mRNA levels of HO-1 and NQO-1 were consistent with their protein expression.
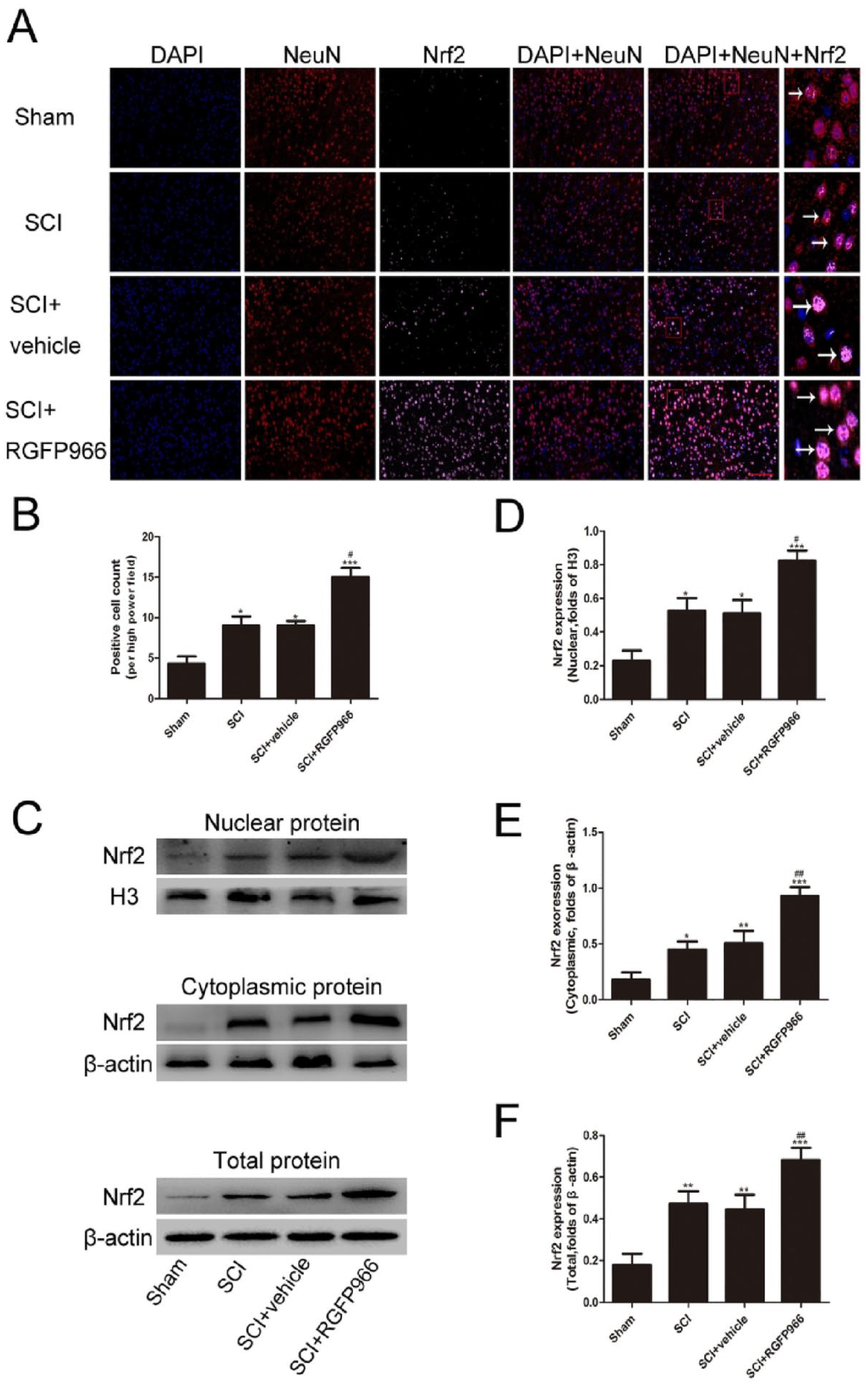
Inhibition of HDAC3 promoted Nrf2 expression and nuclear translocation in the lesion spinal cord after SCI. (A, B) Representative photomicrographs and quantification of Nrf2 immunofluorescent staining. (C-F) Representative Western blot gel images and quantification of Western blot analysis of total, nuclear, and cytoplasmic Nrf2 protein. Data are expressed as the mean ± SEM.
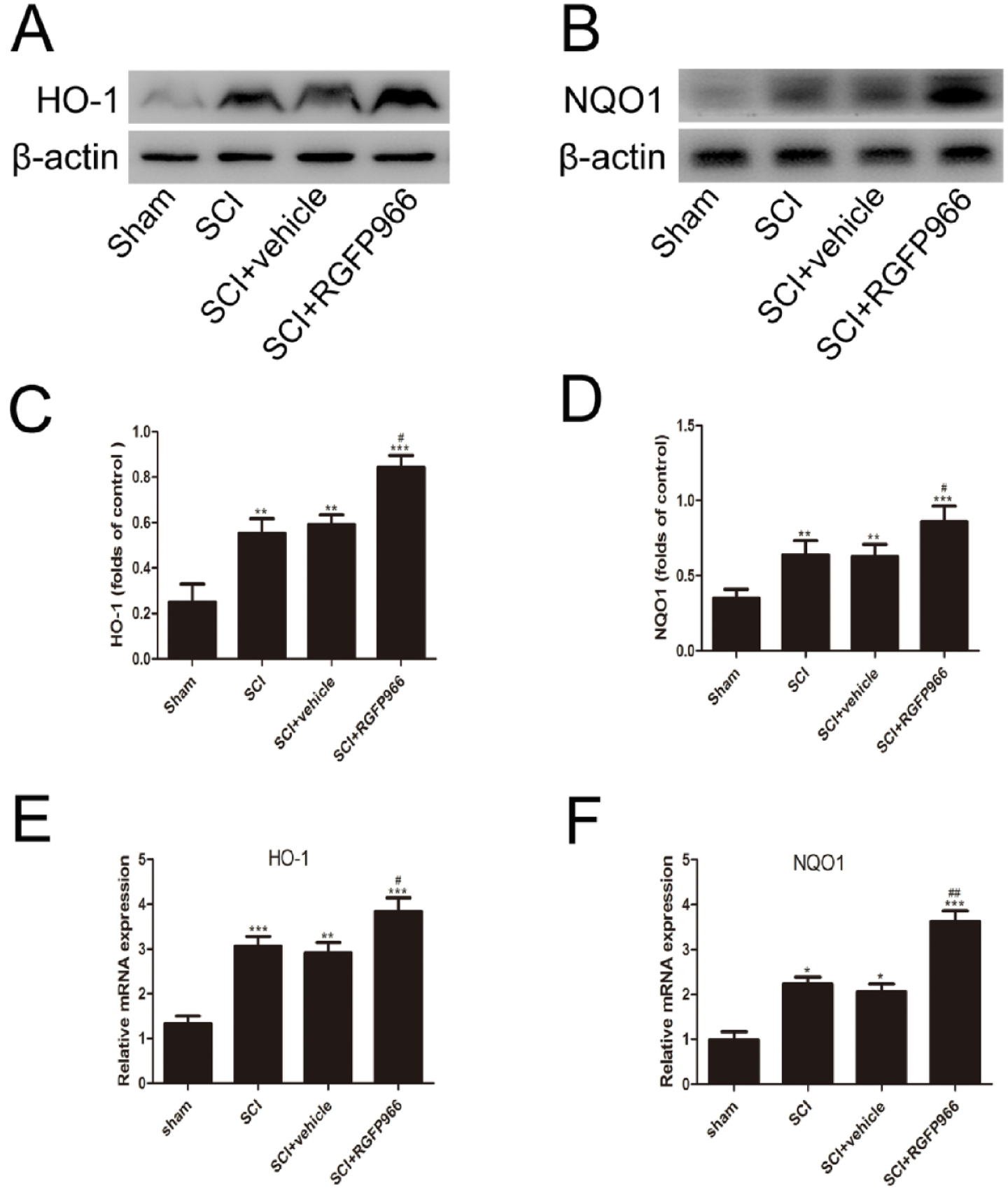
Inhibition of HDAC3 elevated the levels of HO-1 and NQO-1 after SCI. (A-D) Representative Western blot gel images and quantification of Western blot analysis of HO-1 and NQO-1. (E, F) mRNA expression of HO-1 and NQO-1 were analyzed by qRT-PCR. Data are expressed as the mean ± SEM.
Discussion
Secondary injury is a well-known, clinical, nonmechanical damaging factor during the treatment of SCI, and currently, there is no recognized effective treatment for it. 31 Previous study reported that HDAC3 was the main molecular contributor to pathological damage in various neurological disorders, and weakened HDAC3 expression can effectively improve the prognosis of secondary pathological processes.32,33 And, inhibition of HDAC3 would cripple inflammatory response at the injury milieu, leading to neuronal protection preservation after SCI. 34 Our previous study confirmed that HDAC3 expression was increased in lesioned spinal cords, suggesting that histone deacetylation plays an important role in SCI. 16 To confirm whether the activation of HDAC3 has neuroprotective effects, RGFP966, a highly selective inhibitor, was used in this study. The results showed that the inhibition of HDAC3 by RGFP966 attenuated spinal cord edema and BBB permeability, and improved neurological deterioration after SCI. Furthermore, Nissl staining and western blot analysis demonstrated that HDAC3 inhibition reduced neuronal apoptosis and the expression of pro-apoptotic factors, cleaved caspase-3, and Bax. These findings indicate that HDAC3 is a potential therapeutic target against SCI-induced neurological deficits.
It has been demonstrated that microglial activation and its mediation of the inflammatory response plays a crucial role in secondary SCI following SCI. 35 Neuronal necrotic products and various related degradation products stimulate microglia immediately following SCI. The microglia initiate and amplify the inflammatory response, releasing multiple inflammatory factors which cause further damage. 36 Our previous study demonstrated that microglia were activated significantly, and the levels of inflammatory cytokines increased in the spinal cord-damaged region after SCI. 16 In the current study, concomitant with HDAC3 inhibition, microglial activation, inflammatory cytokines release (TNF-α, IL-1β, and IL-6), and neuroinflammation damage were reduced significantly. Altogether, these results support the view that HDAC3 inhibition provides a neuroprotective effect after SCI by inhibiting microglial activation and the subsequent inflammation-induced apoptotic pathways. In addition, these results were in accordance with many previous studies demonstrating that HDAC3 inhibition could protect multiple organs against multifarious noxious stimulation.
It is well known that SIRT1 is a key cytokine in the regulation of oxidation and inflammation in various systems of the body. 37 It is an HDAC that has attracted broad attention across research fields due to its ability to suppress inflammation via its deacetylation activities.38,39 Moreover, there is reliable evidence that SIRT1 is involved in multiple cellular processes and exerts remarkable protective effects against endothelial cell injury, oxidative stress, cell proliferation, and inflammation. 40 Our previous study confirmed that SIRT1 was involved in Beclin-1 deacetylation to ameliorate the inflammatory response associated with neuronal apoptosis. 41 Consistent with previous findings, the results of this study confirmed that HDAC3 inhibition increased SIRT1 expression in lesioned spinal cords. This research confirmed that SCI promoted SIRT1 expression, and the level and nuclear translocation of SIRT1 were further increased when treated with RGFP966. On the basis of these results, we conclude that the protective effects conferred by HDAC3 inhibition are associated with the activation of SIRT1 signaling, and are closely related to the activation of the downstream Nrf2 pathway by SIRT1.
Nrf2, a key antioxidant factor in cellular defense mechanisms, is considered an important downstream target of SIRT1 and increases resistance to noxious stimulation. 42 Under normal circumstances, Nrf2 binds to Kelch-like ECH-associated protein 1 (Keap1), which leads to retrogradation by ubiquitination. In response to noxious stimulation, Nrf2 is dissociated from Keap1 and translocates from the cytosol to the nuclei to subsequently bind to antioxidant response elements (AREs) in the regulatory region of its target genes, thereby activating antioxidant responses by upregulating the level of antioxidant enzymes. 43 In present study, the role of SIRT1/Nrf2 signaling in the protective effects of HDAC3 inhibition was also investigated using a rat model. We found that the expression of SIRT1 and Nrf2 was upregulated in lesioned spinal cords. Interestingly, HDAC3 inhibition further revitalized the activation of SIRT1 and Nrf2, and protected against inflammation as a result, indicating the positive regulatory role of SIRT1 in Nrf2 signaling. NQO1 and HO-1, as Nrf2 targeting genes, were found to be upregulated in the SCI + RGFP966 group, enhancing the effect of post-SCI secondary injury on Nrf2-related protein levels, thereby indicating that HDAC3 inhibition activates the SIRT1/Nrf2 signaling pathway to benefit the anti-inflammatory and antioxidant defense system. These results support the hypothesis that HDAC3 might serve as a key effector molecule in the induction of neuroprotective effects associated with microglial activation and is involved with the downstream SIRT1/Nrf2 signaling pathway.
However, this study has some limitations. Firstly, only preliminary experiments on animals were carried out using specific inhibitors. Further experiments will be necessary to include knockout animal models or cell experiments to assess the overall underlying mechanism. Secondly, the possibility cannot be excluded that other signaling cascades might mediate the effects of HADC3 after SCI. Lastly, the pathway from HADC3 to Nrf2 is complex, and only 1 portion of this pathway as researched. Therefore, further studies are needed to decipher these important mechanisms.
Conclusions
In summary, the results of this study suggest that HDAC3 is implicated in the secondary nonmechanical injury of SCI. The suppression of HDAC3 exerted neuroprotective effects against neuronal apoptosis through suppressing microglia-mediated neuroinflammation after SCI. The protective effects of HDAC3 inhibition might be mediated by activation of SIRT1/Nrf2. On this basis, HDAC3 may become a potential therapeutic target for the treatment of SCI.
Footnotes
Acknowledgements
Sincere appreciation is given to the teachers and our colleagues from The Second Affiliated Hospital of Fujian Medical University, who participated in this study with great cooperation.
Authors Contributions
SBC contributed to the conception, design, and molecular analysis of this study. JFY contributed to the data acquisition and analysis. GZW consulted the literature and revised the manuscript. JNS wrote the manuscript. XRC and WHW provided technical support and revised the manuscript. All authors read and approved the final manuscript.
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the funds for Fujian Province Scientific Foundation (No. 2021J01261) from Dr. Shoubo Chen and Joint Funds for the innovation of Science and Technology, Fujian province (No. 2020Y9033) from Dr. Xiangrong Chen.