
Abstract
Background
Traumatic brain injury (TBI) is a major cause of death and disability worldwide. Oxidative stress, inflammation, and apoptosis are vital pathophysiological features post-TBI.
Objectives
Research has shown that vagus nerve stimulation (VNS) can attenuate oxidative stress in various diseases. However, the critical role of VNS in TBI is still not completely understood. This study investigated the protective effects and potential mechanism of VNS on TBI.
Methods
Male Sprague-Dawley rats were randomized into 3 groups: sham, TBI, and TBI + VNS. The TBI model was induced in rats by the free-fall drop method. The vagal nerve trunk was separated, and VNS was performed after establishing the TBI model.
Results
The results showed that VNS significantly ameliorated tissue damage, neurological deficits, and cerebral edema, compared with the sham VNS group. Additionally, VNS alleviated oxidative stress, inflammation, and apoptosis in the pericontusive cortex of rats after TBI. VNS also significantly suppressed expression of the nuclear factor-κB (NF-κB) protein in the nucleus and activation of the nucleotide-binding domain–like receptor protein 3 (NLRP3) inflammasome.
Conclusions
Taken together, the present study indicates that VNS may attenuate brain damage after TBI by inhibiting oxidative stress, inflammation, and apoptosis, possibly through the NF-κB/NLRP3 signaling pathway.
Keywords
Introduction
Traumatic brain injury (TBI) is a major cause of death and disability in adolescents worldwide. 1 The morbidity of TBI is still rising, even in developed countries, and has gradually become a silent epidemic. Published data reported that 1.7 million people undergo TBI annually in the United States, approximately double that in China. 2 Numerous untreated brain injuries are accompanied with complications such as posttraumatic stress disorder, cognitive or behavioral impairment, epileptic seizures, chronic encephalopathy, and neurodegenerative disease. 3 TBI is initiated by mechanical injury, followed by a range of secondary damage cascades. The secondary pathological processes include oxidative stress, inflammation, apoptosis, mitochondrial dysfunction, impaired calcium and iron homeostasis, and excitotoxic damage, all of which may require clinical intervention.4,5 Although several treatments, such as progesterone, minocycline, melatonin, statins, and mesenchymal stem cells, exhibited encouraging results in preclinical studies and early clinical trials, none has advanced to phase III clinical trials.6 -9 Therefore, development of novel, effective adjuvant treatments to ameliorate neuronal injury and improve functional outcomes after TBI are needed.
Oxidative stress plays a pivotal role in brain injuries and may be a therapeutic target for TBI. 10 After traumatic injury, oxidative stress induces the release of proinflammatory factors and exaggerates inflammation through nuclear transcription factor-κB (NF-κB) activation. 11 Among the TBI-induced proinflammatory cytokines, interleukin-1β (IL-1β) triggers the TBI-induced inflammatory cascade. 12 The nucleotide-binding domain (NOD)-like receptor protein 3 (NLRP3) inflammasome, one of the NOD-like receptors, can regulate the secretion of IL-1β.13,14 Moreover, the NLRP3 inflammasome is also involved in other neurological diseases such as Alzheimer’s disease, pneumococcal meningitis, and Huntington’s disease.15-17 Therefore, NLRP3 may be an effective target to block the development of neuroinflammation in TBI.
Vagus nerve stimulation (VNS) exerts anti-inflammatory effects in a variety of diseases 18 and is considered a safe and effective therapy for refractory depression, schizophrenia, somatoform disorder, refractory epilepsy, and inflammatory bowel disease.19-23 Our previous study also revealed that VNS improved consciousness recovery from TBI. 24 However, the effects and potential mechanism of VNS in TBI are still not well-understood. Therefore, in the present study, we examined the protective effects of VNS on TBI-induced brain injury and discussed whether the NF-κB/NLRP3 signaling pathway was involved in these effects.
Methods
Animals and Experimental Groups
Male Sprague-Dawley rats (aged 6-8 weeks, weighing 250-300 g) were purchased from the Hunan SJA Experimental Animals Company (Changsha, Hunan Province, China; License No. SCXK [Xiang] 2016-0002). Rats were maintained at 23 ± 2 °C with a 12-hour light/dark cycle and free access to food and water. The animals were adapted to laboratory conditions for 7 days before the experiment. Rats were divided randomly into 3 groups: sham operation (sham group; n = 30, 278.5 ± 14.6 g), TBI with sham VNS (TBI group; n = 30, 276.6 ± 17.3 g), and TBI with VNS (TBI + VNS group; n = 30, 269.5 ± 18.7 g).
TBI Model
The TBI model was established as described in our published study. 25 Briefly, rats were anesthetized, a midline longitudinal incision was made in the scalp, and the skin was retracted to expose the skull. Subsequently, the rats were fixed in a stereotaxic instrument with 2 ear bars and a nose clamp. A cross was marked 2 mm left of the midline and 1 mm anterior to the coronal suture using a needle. Then, a 2 cm diameter and 350 g cylindrical impact hammer (Guangyuan Forging) was dropped onto the marked cross from a height of 40 to 45 cm, resulting in a fracture of the skull (Figure 1A). The incision was disinfected and sutured, and the rats were then kept in clean cages. Rats in the sham group underwent a similar surgical procedure as the TBI group, but the weight-drop test was not conducted.
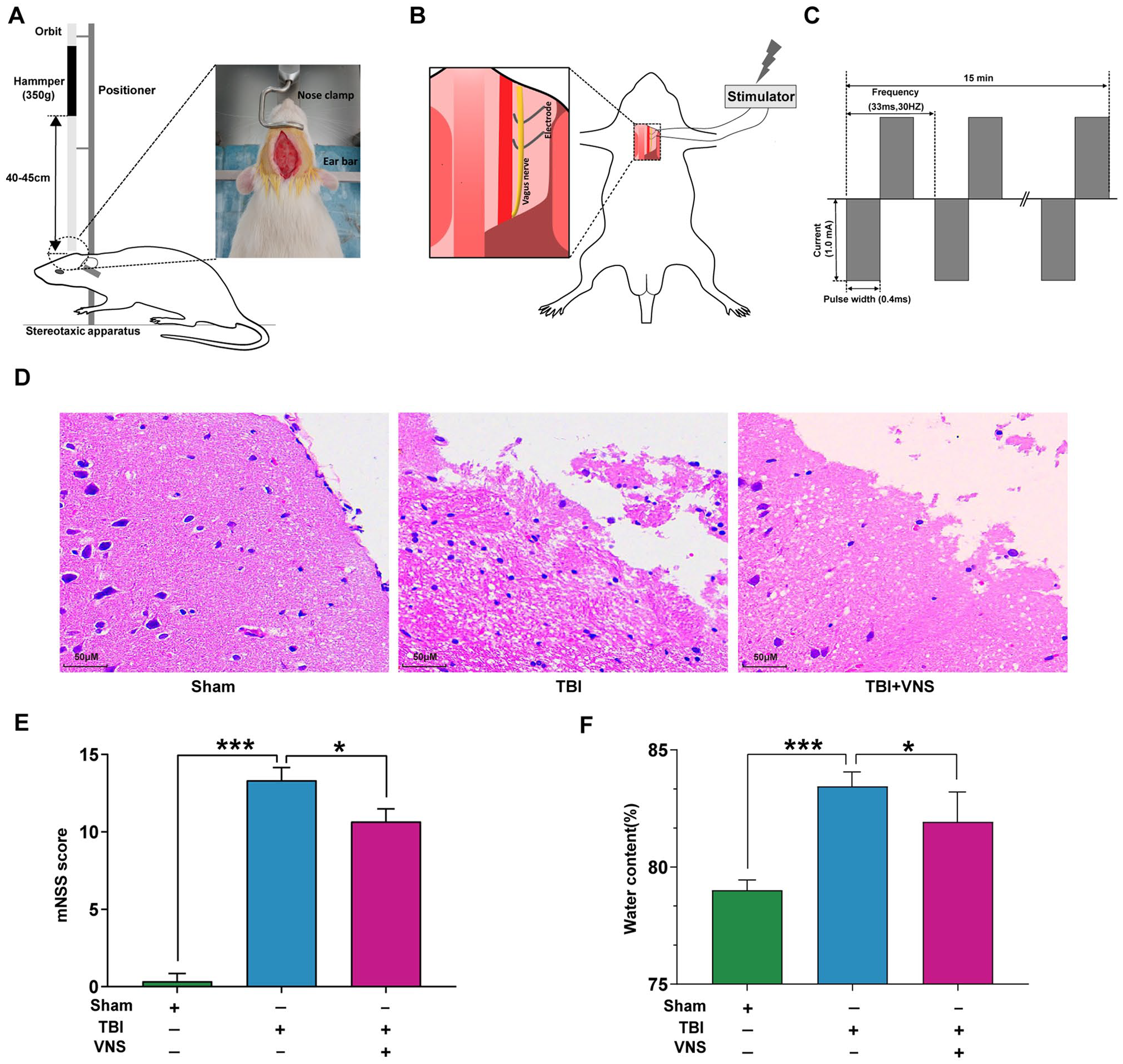
Vagus nerve stimulation (VNS) alleviates tissue damage, neurological deficits, and cerebral edema. (A) Construction of the traumatic brain injury (TBI) model in rats. (B) VNS in rats. (C) Parameters and process for the stimulation. (D) Representative hematoxylin and eosin staining images of cortex tissues show necrotic areas in the 3 groups. (E) Neurological function was analyzed by modified neurological severity scores (mNSS) at 24 hours after TBI. (F) The brain water content of each group was detected 24 hours post-TBI (*P < .05, **P < .01, ***P < .001).
VNS
VNS was performed 30 minutes after the TBI procedure. A small incision was made on the left ventral side of the neck adjoining the midline to approach the left vagus nerve at the cervical level. We performed a blunt dissection of the subcutaneous fat, salivary glands, sternohyoid, and sternocleidomastoid, and cut the carotid sheath open to expose the vagus nerve. A 5 mm segment of the left vagus nerve was separated and attached to an electrode (Figure 1B). To ensure good contact between the electrode and vagus nerve, an ohmmeter was used. Ohmmeter probes were connected to the electrode and electrical stimulator, and the electrode was then attached to the vagus nerve. The electrode was composed of a pair of Teflon-coated silver hooks. To avoid short circuit, water was dried with gauze. Rats were treated with VNS using a low-frequency electrical stimulator (ES-420; ITO Physiotherapy & Rehabilitation) with the following parameters: frequency, 30 Hz; current, 1.0 mA; pulse width, 0.5 ms; total stimulation time, 15 minutes 24 (Figure 1C). Following surgery, each animal received an intramuscular injection of gentamicin (0.1 mL/100 g body weight). Rats in the sham and TBI groups underwent treatments that were similar to those in the stimulated groups, but without electrical stimulation.
Modified Neurological Severity Score (mNSS)
The mNSS was determined 24 hours post-TBI to evaluate neurological function. The mNSS test includes 10 different parts that assess the motor (muscle status, abnormal movement), sensory (visual, tactile, and proprioceptive), balance, and reflex functions of rats. 26 Rats were scored from 0 to 18 (0 = normal function; 18 = maximal deficit). One point was scored for each abnormal behavior or for the absence of a tested reflex. All neurobehavioral tests were carried out by 2 investigators (Kang and Dong) who were blinded to the experimental groups. Any disagreements were solved through consulting to reach consensus by the third researcher (Feng).
Brain Water Content
Following the experiment, rats were sacrificed by decapitation. Whole brain tissues (n = 6, ie, 6 out of 30 rats from each group) were removed and divided into 2 hemispheres along the midline. The olfactory bulb, meninges, lower brain stem, and cerebellum were subsequently dissected, washed with normal saline, and dried with filter paper. The samples were then immediately weighed in the glass vail to obtain the wet weight. Subsequently, the glass vails with the samples were placed in the oven at 100 °C for 24 hours and weighed again to determine the dry weight. The percentage of water in each brain was calculated using the following formula: brain water content (%) = [(wet weight − dry weight)/wet weight] × 100%.
Hematoxylin and Eosin Staining
At 24 hours postinjury, the rats (n = 6) were decapitated after induction of anesthesia by ether, and the brain tissues were harvested. The isolated samples were fixed with 4% paraformaldehyde for 24 hours at room temperature, and dehydrated using graded alcohol with a series of 70%, 80%, 90%, 95%, and 100% ethanol, and finally embedded in paraffin, and cut into 5-µm-thick slices. Then, hematoxylin and eosin staining of the paraffin-embedded brain tissues was performed. Briefly, sections were dewaxed and rehydrated with graded ethanol, stained with hematoxylin for 6 minutes, then stained with eosin for 3 minutes. The slides were then examined using a light microscope.
Biochemical Assays
The sample from each rat was divided into 4 portions, and then the bioassay was performed using 4 different testing kits (n = 6). We employed the 2-thiobarbituric acid method to measure malondialdehyde (MDA) and a colorimetric method to measure glutathione (GSH) levels in cortex homogenates. The xanthine oxidase and ammonium molybdate spectrophotometric methods were used to detect superoxide dismutase (SOD) and catalase (CAT) activities, respectively, of cortex homogenates. These commercial kits were all purchased from Nanjing Jiancheng Bio-Engineering Institute.
Terminal Deoxynucleotidyl Transferase (TdT) dUTP Nick-End Labeling (TUNEL) Staining
Paraffin-embedded sections were stained with a TUNEL assay kit (Roche) to detect apoptotic cells in the cortex tissue according to the manufacturer’s protocol. TUNEL-positive apoptotic nuclei stained red, and TUNEL-negative nuclei stained blue. The apoptotic index was calculated as a ratio of the number of TUNEL-positive nuclei to total nuclei. Fluorescence images were assessed on an Olympus IX71 inverted microscope system. All examinations were conducted by 2 investigators who were blinded to the grouping.
Western Blot
Pericontusive cortex (3 × 3 × 5 mm (width × depth × length) surrounding the edge of the lesion) proteins were extracted (n = 6), and protein levels of Bcl-2-associated X protein (Bax), B-cell lymphoma 2 (Bcl-2), NF-κB, NLRP3, apoptosis-associated speck-like protein (ASC), and caspase-1 were measured by western blotting. Protein concentrations from tissue lysates were measured using the bicinchoninic acid method (cat. no. PA115; Tiangen Biotech). Primary antibodies for western blotting were anti-Bax (1:1000, Abcam, ab32503), anti-Bcl-2 (1:1000, Abcam, ab59348), anti-NF-κB (1:1000, Cell Signaling Technology, 8242), anti-NLRP3 (1;500, Abcam, ab214185), anti-ASC (1:500, Abcam, ab175449), anti-caspase-1 (1:1000, Novusbio, NB100-56565), and anti-GAPDH (1:2000, Proteintech, 60004-1-lg). Following sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), the protein from each sample was transferred to the polyvinylidene fluoride (PVDF) membrane. To make the primary antibodies bind to the secondary antibodies, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (1:5000, Zsgb-Bio) for 60 minutes at room temperature. Western blot results were analyzed using Image Lab Software (version 3.0).
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA from pericontusion traumatic brain tissues (n = 6) was extracted using TRIzol reagent (Invitrogen). The RNA concentration was measured using an ultraviolet spectrophotometer (Shanghai Precision Scientific Instrument Corp). Then, total RNA was used for reverse transcription to synthesize cDNA using a commercial kit (TransGen Biotech) according to the manufacturer’s instructions. QRT-PCR was performed using the SYBR Green PCR Master Mix (TransGen Biotech). The reactions were performed on a StepOne Real-Time PCR System (Thermo Fisher Scientific). The amplification program was performed as follows: denaturation at 95 °C for 15 minutes, followed by 40 cycles including annealing for 10 seconds at 95 °C and extension for 32 seconds at 60 °C. The melting curve started at 95 °C for 15 seconds, followed by 60 °C for 1 minutes, and ended with 15 seconds at 95 °C. The mRNA expression was quantified using the 2−ΔΔCt method. The primers used are presented in Table 1.
The Primer Sequences for PCR Amplification.

Abbreviations: PCR, polymerase chain reaction; Tm, melting temperature; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; Bcl-2, B-cell lymphoma 2; NLRP3, nucleotide-binding domain (NOD)-like receptor protein 3; IL-1β, interleukin-1β; TNF-α, tumor necrosis factor-α.
Immunohistochemical Staining
The brain tissue samples used for immunohistochemical staining were obtained from the rats of the same subgroup that was also used for hematoxylin and eosin staining (n = 6). Protein levels of IL-18 and IL-1β were detected using immunohistochemical staining in paraffin-embedded sections. The sections were cut into 5-µm-thick slices, deparaffinized in xylene, dehydrated in a graded series of 100%, 95%, 85%, and 80% ethanol, and subjected to antigen retrieval in citrate buffer (pH 6.0) for 30 minutes in a 37 °C chamber. Subsequently, the sections were rinsed thrice for 5 minutes each and then incubated with normal goat serum for 20 minutes. Then, the sections were incubated overnight at 4 °C with rabbit anti-IL-18 (1:2000, Abcam, ab71495) and anti-IL-1β (1:50, Abcam, ab9787). The secondary antibody was goat anti-rabbit (1:5000, Zsgb-Bio). The slides were observed using a light microscope. The positive staining area was quantified using ImageJ software (US National Institutes of Health).
Statistical Analyses
Continuous data (normal distribution) are presented as means ± standard deviation. Significant differences in these data between the groups were determined using a one-way analysis of variance with Tukey’s post hoc analysis. For the ranked neurological severity score (mNSS) data (nonnormal distribution), significant differences between the groups were assessed using the Kruskal-Wallis test. Statistical analyses were conducted using GraphPad Prism 8.0 software. P < .05 was considered statistically significant.
Ethics Approval
The experimental protocol of this study was approved by the Research Committee of the First Affiliated Hospital of Nanchang University, Nanchang, China (Approval No. 003) in January 2016. The rats were sacrificed by decapitation under ether anesthesia, and the brain tissue was obtained quickly within 2 minutes.
Results
VNS Reduced Tissue Damage, Neurological Deficits, and Brain Water Content
The sham group exhibited clear and dense brain structures. Abundant neuronal structures were normal and arranged uniformly. In contrast, the TBI group displayed loosened structures, and neurons had extensive vacuolar changes and cellular necrosis. VNS treatment significantly alleviated brain edema and necrosis in lesioned brain tissues (Figure 1D).
We conducted the mNSS test to evaluate the effects of VNS on neurological deficits at 24 hours post-TBI. Compared to the sham group, the TBI group showed higher scores. However, VNS treatment significantly lowered the mNSS scores, suggesting a protective role of VNS in neurological injury (P < .001; Figure 1E). We further explored the therapeutic effects of VNS treatment on TBI-induced brain edema. The brain water content in the TBI group was significantly higher than that in the sham group, while VNS treatment significantly ameliorated brain edema (degree of freedom [df] = 2, 15, F = 41.55, P < .001; Figure 1F).
VNS Ameliorated Oxidative Stress Damage in the TBI Model
We next explored the effect of VNS on oxidative stress in the TBI model. The level of MDA, a marker of oxidative stress, was dramatically elevated in the TBI group compared with the sham group, and VNS treatment significantly reduced this level (df = 2, 15, F = 117.0, P < .001; Figure 2A). The level of GSH, a powerful antioxidant, was significantly decreased in TBI model brains when compared with the sham group, whereas VNS increased the GSH level compared with that in the TBI alone group (df = 2, 15, F = 11.67, P < .001; Figure 2B). The activities of SOD and CAT, 2 important antioxidant enzymes, were noticeably decreased in the TBI group compared with the sham group, while their activities were enhanced after VNS treatment (SOD, df = 2, 15, F =13.34, P < .001, Figure 2C; CAT, df = 2, 15, F= 26.08, P < .001, Figure 2D).
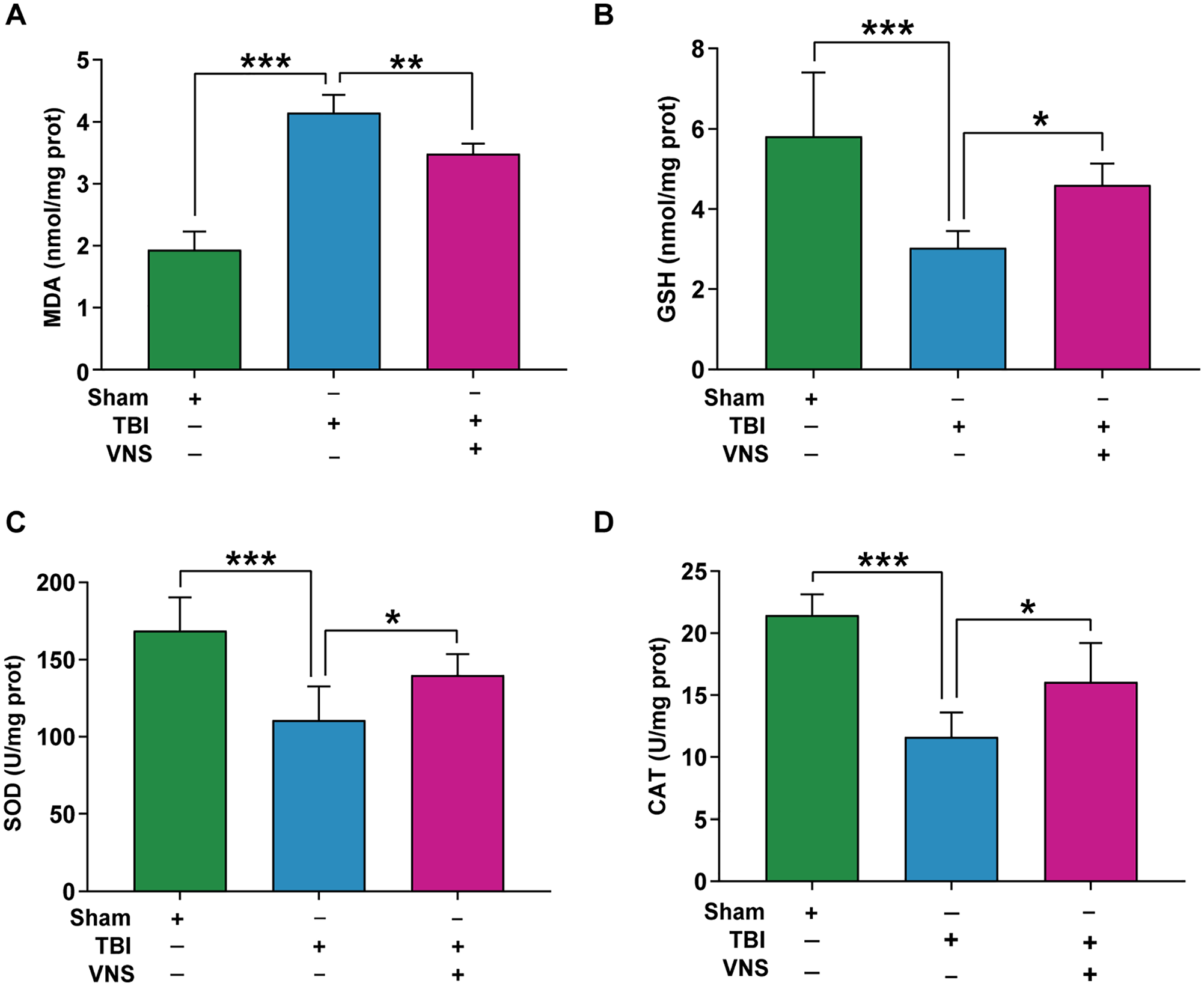
Vagus nerve stimulation (VNS) attenuates oxidative stress after traumatic brain injury (TBI). The effect of VNS on the (A) malondialdehyde (MDA), (B) glutathione (GSH), (C) superoxide dismutase (SOD), (D) catalase (CAT). Results are expressed as means ± standard deviation (SD; *P < .05, **P < .01, ***P < .001).
VNS Inhibited Neuronal Apoptosis in the TBI Model
We explored the effect of VNS on neuronal apoptosis in the TBI model. TUNEL staining revealed that the number of apoptotic neurons was significantly elevated in the TBI group, but was reduced in the TBI + VNS treatment group (df = 2, 15, F = 80.04, P < .001; Figure 3A). Bax and Bcl-2 are key pro- and anti-apoptotic molecules, respectively. The protein and mRNA levels of Bax significantly increased in the TBI group, while they decreased after VNS treatment (protein: df = 2, 15, F = 58.68, P < .001, Figure 3B; mRNA: df = 2, 15, F = 54.42, P < .001, Figure 3C). Inversely, the protein and mRNA levels of Bcl-2 significantly decreased in the TBI group, and the protein increased after VNS treatment (protein: df = 2, 15, F = 128.7, P < .001, Figure 3D; mRNA: df = 2, 15, F = 63.63, P < .001, Figure 3E).
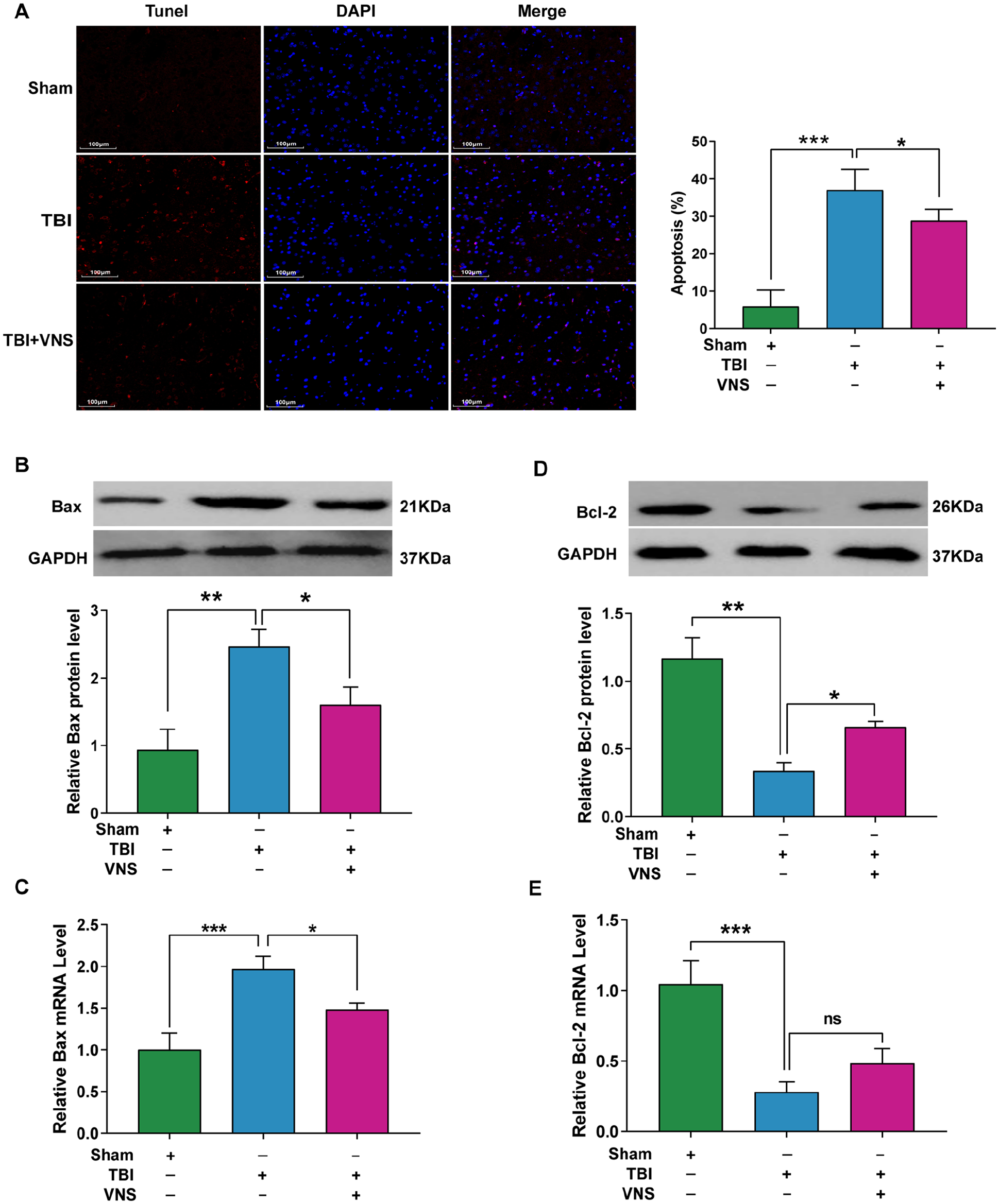
Vagus nerve stimulation (VNS) inhibits neuronal apoptosis by regulating Bcl-2 (B-cell lymphoma 2) and Bax levels. (A) Deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL; red) and 4′,6-diamidino-2-phenylindole (DAPI; blue) staining in pericontusive cortex after traumatic brain injury (TBI). Percentages of TUNEL-positive cells are shown. (B) Relative protein levels of Bax. (C) Relative mRNA levels of Bax. (D) Relative protein levels of Bcl-2. (E) Relative mRNA levels of Bcl-2. Results are expressed as means ± standard deviation (SD; *P < .05, **P < .01, ***P < .001).
VNS Suppressed NF-κB/NLRP3 Inflammasome Activation in the TBI Model
NF-κB/NLRP3 signaling pathway plays a crucial role in oxidative stress damage. Compared with the sham group, the protein level of NF-κB significantly reduced in cytoplasm and elevated in the nucleus under TBI conditions, while VNS treatment reversed these changes (cytoplasm: df = 2, 15, F = 120.2, P < .001, Figure 4A; nucleus: df = 2, 15, F = 10.54, P = .0014, Figure 4B). The NLRP3 inflammasome consists of NLRP3, ASC, and caspase-1 molecules. The protein and mRNA levels of NLRP3 significantly increased in the TBI group but decreased after VNS treatment (protein: df = 2, 15, F = 20.44, P < .001, Figure 4C; mRNA: df = 2, 15, F = 16.29, P < .001, Figure 4D). The levels of ASC and caspase-1 proteins increased significantly in the TBI group compared with the sham group, whereas their protein levels decreased after VNS treatment (ASC: df = 2, 15, F = 20.44, P < .001, Figure 4E; caspase-1: df = 2, 15, F = 16.29, P < .001, Figure 4F).
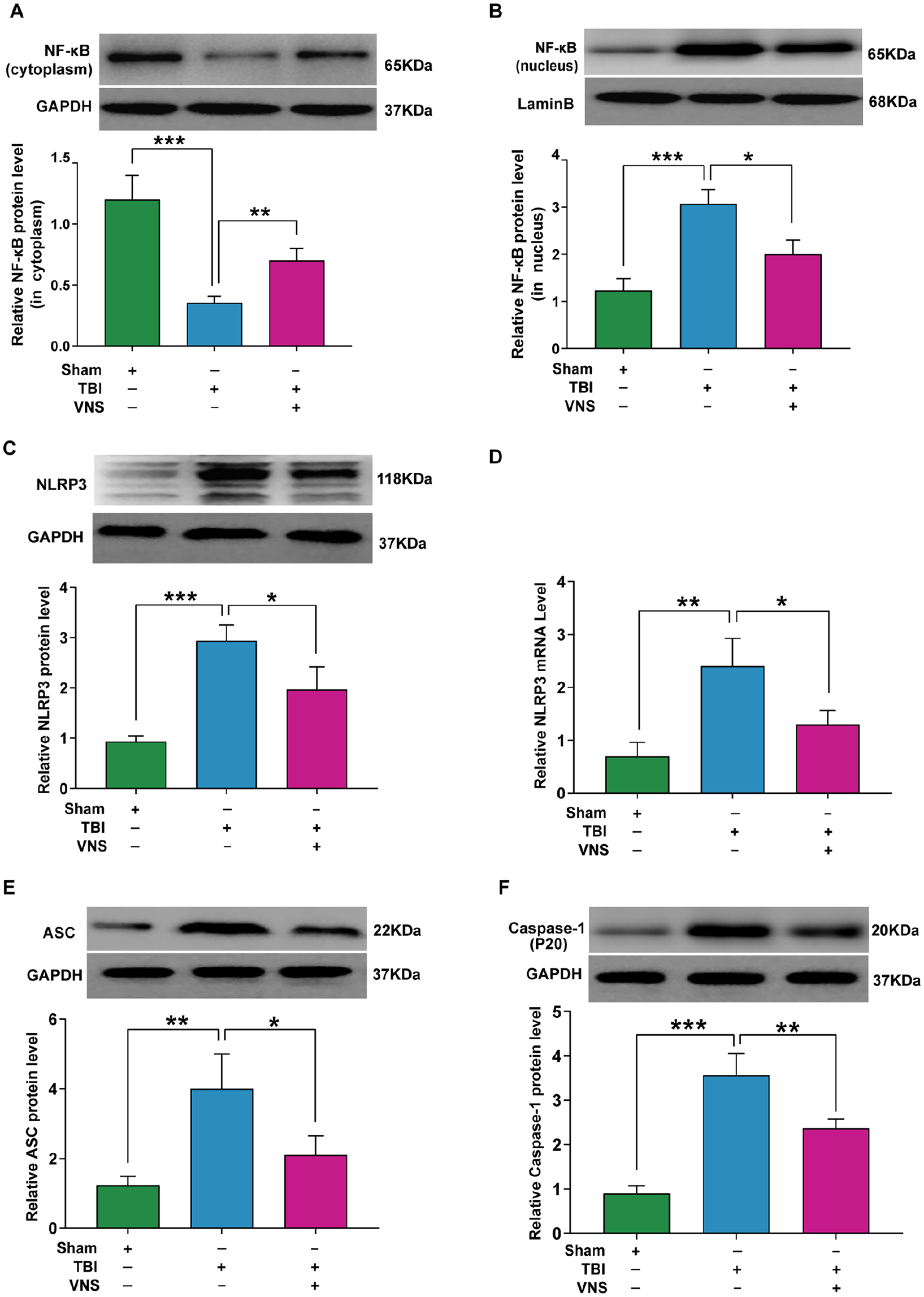
Vagus nerve stimulation (VNS) inhibits NF-κB (nuclear factor kappa B) and nucleotide-binding domain (NOD)-like receptor protein 3 (NLRP3) inflammasome activation after traumatic brain injury (TBI). (A) NF-κB protein level in the cytoplasm. (B) NF-κB protein level in the nucleus. (C) VNS significantly downregulates the protein levels of NLRP3. (D) VNS significantly downregulates the mRNA levels of NLRP3. (E) VNS significantly downregulates the protein expression of apoptosis-associated speck-like protein (ASC). (F) VNS significantly downregulates the protein expression of caspase-1. Results are expressed as mean ± standard deviation (SD; *P < .05, **P < .01, ***P < .001).
VNS Suppressed the Production of Proinflammatory Cytokines in the TBI Model
We detected the effects of VNS on the levels of proinflammatory cytokines in the TBI model. Immunohistochemical staining showed that IL-1β and IL-18 protein levels were consistent with their mRNA levels (IL-1β: df = 2, 15, F = 11.10, P = .0011; IL-18: df = 2, 15, F = 23.20, P < .001; Figure 5A). We also investigated the mRNAs of proinflammatory cytokines. Compared with the sham group, IL-1β, IL-18, IL-6, and tumor necrosis factor-α (TNF-α) mRNA levels increased in the TBI group, while VNS treatment reduced the levels of these proinflammatory cytokines. In addition, immunohistochemical staining showed that IL-1β and IL-18 protein levels were consistent with their mRNA levels (IL-1β: df = 2, 15, F = 22.16, P = 0.0011; IL-18: df = 2, 15, F = 22.47, P < .001; IL-6: df = 2, 15, F = 29.85, P = .0011; TNF-α: df = 2, 15, F = 20.99, P < .001; Figure 5B).
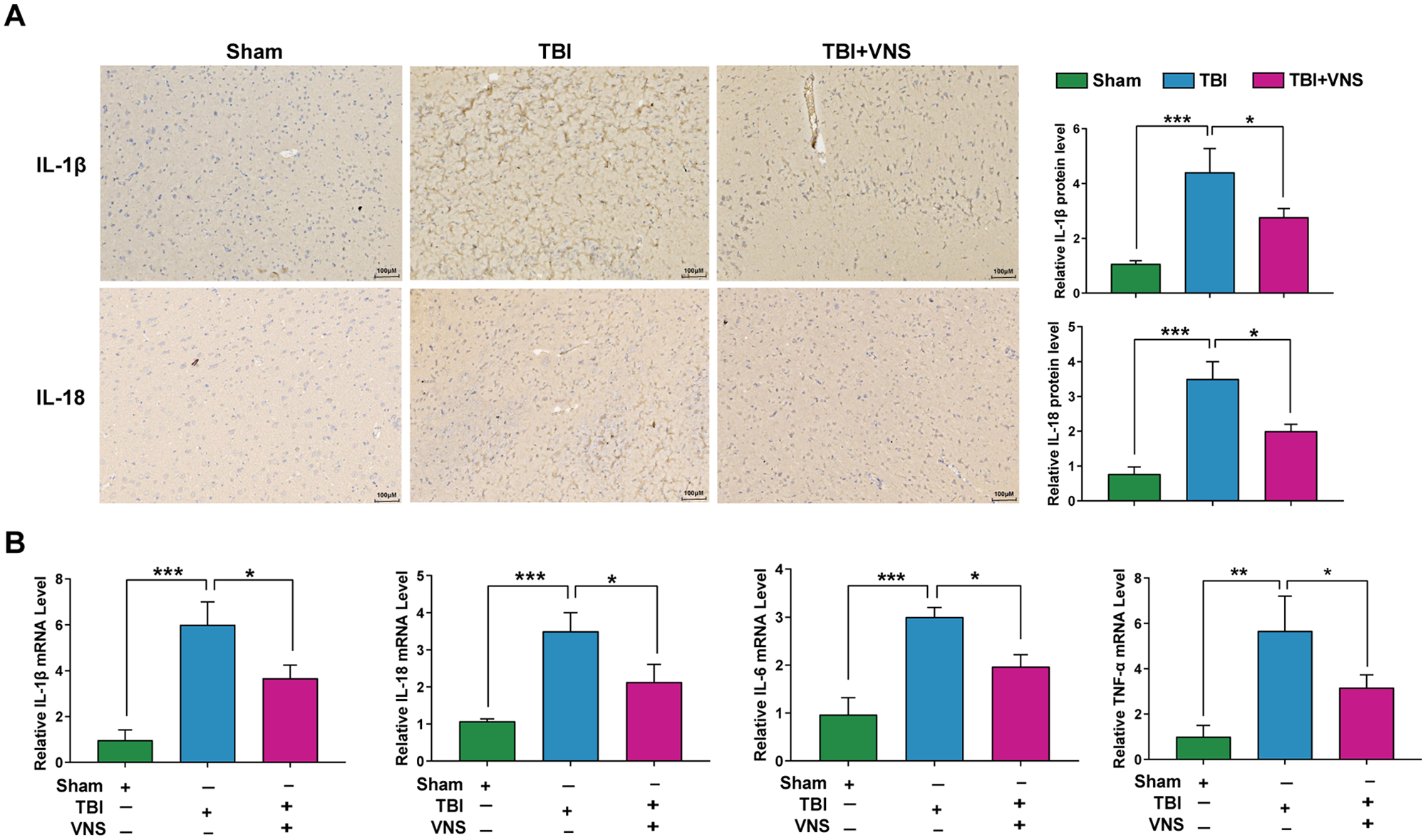
Vagus nerve stimulation (VNS) mitigates inflammation after traumatic brain injury (TBI). (A) Representative immunohistochemical staining and relative protein expression of interleukin (IL)-1β and IL-18. (B) Relative mRNA levels of IL-1β, IL-18, tumor necrosis factor (TNF)-α, and IL-6. Results are expressed as mean ± standard deviation (SD; *P < .05, **P < .01, ***P < .001).
Discussion
In the current study, we found that VNS attenuated early TBI by suppressing oxidative stress, inflammation, and apoptosis. In addition, we found that VNS markedly inhibited the NF-κB/NLRP3 pathway in the brain injury zone. Our results demonstrate that VNS may exert its neuroprotective role in the TBI model of brain injury by regulating NF-κB/NLRP3 signaling (Figure 6).
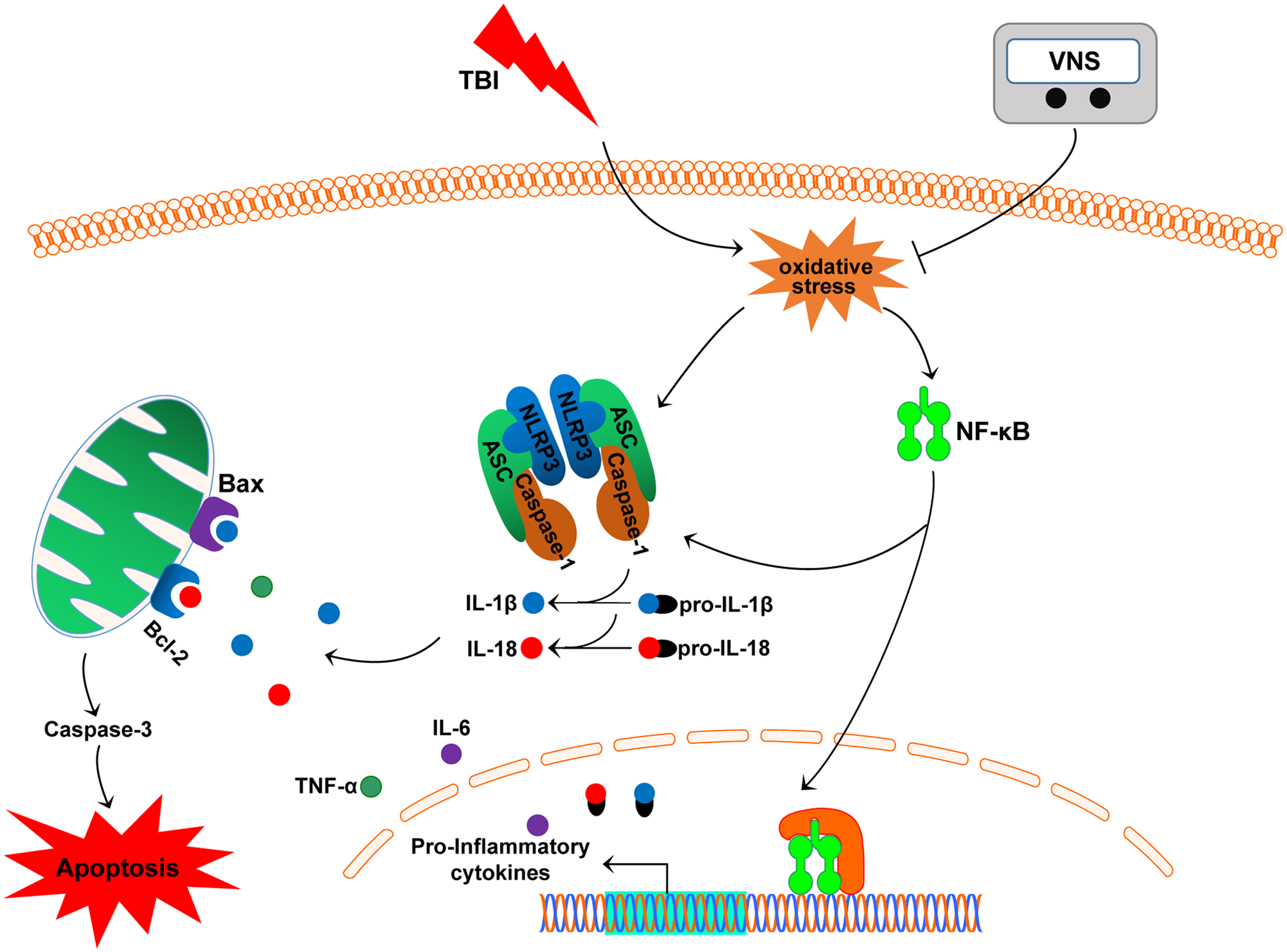
Schematic diagram depicting the potential mechanism and protective effects of vagus nerve stimulation (VNS) on traumatic brain injury (TBI). VNS exerts neuroprotective effects in TBI model rats through inhibition of oxidative stress, inflammation, and apoptosis mediated by the NF-κB/nucleotide-binding domain (NOD)-like receptor protein 3 (NLRP3) signaling pathway.
VNS is a neuromodulation technique used clinically for several neurological diseases such as refractory epilepsy and depression. 27 Several studies have examined the effect of VNS post-TBI. In our previous study, we investigated the specific roles of VNS in comatose rats after TBI. The results indicated that orexin-A, an important neuropeptide that participates in the sleep/wake cycle, might be involved in the wake-promoting effects of VNS through binding and activation of orexin receptor type 1 (OXR1). 24 Neese et al found that VNS protected glutamic acid decarboxylase-positive cortical cells from death after TBI and increased glutamic acid decarboxylase-positive cell counts in the hippocampal hilus of the injured brain. 28 VNS can also enhance motor and cognitive recovery, reduce blood-brain barrier breakdown, and confer neuroprotective effects.29-32 Ghrelin may also play an important role in the neuroprotective effects of VNS. 33 VNS may enhance neural plasticity following TBI by enhancing the release of norepinephrine. 29 However, most of these studies did not investigate the potential molecular mechanism of VNS in TBI.
Oxidative stress results from a disturbance in reactive oxygen species (ROS, such as singlet oxygen, hydrogen peroxide, hydroxyl radicals, and superoxide) production and/or degradation leading to an oxidant-antioxidant imbalance, and accompanies the primary mechanical impact in the TBI model. Oxidative stress can exaggerate other secondary injuries such as inflammation and apoptosis.34,35 Brain tissue is enriched with fatty acids that are prone to peroxidation, and has limited antioxidant defenses. 36 In TBI models, antioxidant defense systems against ROS production fail, which causes protein oxidation, inhibits the mitochondrial electron transport chain, and cleaves DNA. 37 Consequently, brain tissue may suffer damage due to excessive oxidative stress. In the present study, we also investigated the state of oxidative stress induced by TBI and our results were consistent with previous findings.
MDA has been considered an index of lipid peroxidation. 38 The important enzymatic antioxidants, including CAT, SOD, and GSH, prevent the brain from oxidative injury. 39 The present results suggest that VNS can prevent the decreased activities of CAT and SOT, and level of GSH, suggesting that the oxidative injury induced by TBI was drastically reduced. Compared with the TBI group, the MDA level was markedly decreased after VNS treatment, demonstrating the defensive role of VNS in TBI.
Oxidative stress can trigger various deleterious effects and activate NF-κB. 40 The NF-κB signaling pathway plays a crucial role in inflammation-related diseases by inducing the transcription of proinflammatory cytokine genes and causing a positive feedback loop. 41 Within 24 hours post-TBI, the blood-brain barrier is dysfunctional, and circulating neutrophils, monocytes, and lymphocytes infiltrate the damaged brain parenchyma. 42 Evidence shows that early inflammation causes brain injury and results in neuronal lesions, 43 suggesting that inflammation is a vital mediator of secondary brain injury. The expression of NF-κB in the cytoplasm and nucleus was used to detect the activation of NF-κB, as only the transfer of NF-κB from the cytoplasm to the nucleus results in its role as a transcription factor. In our present study, we detected the expression of NF-κB in the cytoplasm and nucleus to evaluate its transcriptional activity. It was found that VNS treatment inhibited NF-κB activation and the secretion of proinflammatory cytokines. Thus, these results again support the anti-inflammatory effects of VNS.
The NLRP3 inflammasome, a downstream mediator of NF-κB, is a member of the recognition receptor family involved in the innate immune response and plays a role in brain damage post-TBI. 44 The NLRP3 inflammasome contains a NLRP3 scaffold, ASC adaptor, and procaspase-1. After it is activated, procaspase-1 is cleaved to caspase-1, triggering the release of IL-1β and IL-18. Activation of the NLRP3 inflammasome directly depends on self-activation and/or indirectly depends on the NF-κB pathway. 45 Secondary brain injury exhibits characteristic neuronal and oligodendrocyte apoptosis. 46 Apoptosis is dependent on activation of caspases-3, -8, and -9 through the exogenous death receptor or endogenous mitochondrial pathways. 47 Additionally, pro- and anti-apoptotic factors, such as Bax and Bcl-2, are involved in apoptosis. Studies have reported that the expression of Bax is significantly upregulated, and the expression of Bcl-2 is downregulated in the TBI model. 48 The NLRP3 inflammasome is a multiprotein oligomer activated by NF-κB and promotes the secretion of IL-1β and IL-18. These cytokines induce pro-inflammatory cell death by activating Bax. Therefore, activation of NF-κB and its subsequent stimulation of the NLRP3 inflammasome, and release of inflammatory cytokines provoked apoptosis in the TBI rats. Our study is consistent with a series of previous studies that NF-κB/NLRP3 inflammasome signaling and apoptosis are involved in the pathophysiological mechanism of TBI. Importantly, we found that VNS inhibited the activation of NF-κB/NLRP3 inflammasome signaling and apoptosis. Collectively, these results demonstrate that VNS may inhibit apoptosis induced by the activation of NF-κB/NLRP3 signaling pathway post-TBI.
There are several limitations in this study. First, we only explored the neuroprotective effects of VNS in early brain injury post-TBI; thus, the effects of VNS on the chronic stage of brain damage post-TBI need to be investigated. Second, age, gender, weight, and other features are associated with the prognosis of TBI, but we only explored the effect of VNS in rats of similar age, gender, and weight. Third, involvement of the NF-κB/NLRP3 inflammasome pathway in VNS treatment for this TBI model requires further study through loss-of-function and gain-of-function research. In addition, there are several other possible mechanisms that need to be investigated in future studies.
Conclusions
These results demonstrate that VNS exerts neuroprotective effects in a TBI rat model through inhibition of oxidative stress, inflammation, and apoptosis mediated by the NF-κB/NLRP3 signaling pathway. The results of this study shed light on the potential therapeutic use for VNS in TBI.
Footnotes
Authors’ Note
The data used to support the findings of this study are available from the corresponding author on request.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Support was provided by grants from the National Natural Science Funds of China (No. 81860409 and No. 81660382), the Youth Foundation of Science and Technology Research of Jiangxi Educational Committee (No. GJJ190125), and Graduate Students Innovation Fund Project in Jiangxi Province (No. YC2019-B036).