
Abstract
Activity-dependent treatments to enhance peripheral nerve regeneration after injury have shown great promise, and clinical trials implementing them have begun. Success of these treatments requires activity-dependent release of brain-derived neurotrophic factor (BDNF). A single nucleotide polymorphism (SNP) in the bdnf gene known as Val66Met, which is found in nearly one third of the human population, results in defective activity-dependent BDNF secretion and could impact the effectiveness of these therapies. Here, we used a mouse model of this SNP to test the efficacy of treadmill exercise in enhancing axon regeneration in animals both heterozygous (V/M) and homozygous (M/M) for the SNP. Axon regeneration was studied 4 weeks after complete transection and repair of the sciatic nerve in both male and female animals, using both electrophysiological and histological outcome measures. Regeneration was enhanced significantly without treatment in V/M mice, compared with wild type (V/V) controls. Unlike V/V mice, treatment of both V/M and M/M mice with treadmill exercise did not result in enhanced regeneration. These results were recapitulated in vitro using dissociated neurons containing the light-sensitive cation channel, channelrhodopsin. Three days after plating, neurites of neurons from V/M and M/M mice were longer than those of V/V neurons. In neurons from V/V mice, but not those from V/M or M/M animals, longer neurites were found after optogenetic stimulation. Taken together, Met-carriers possess an intrinsically greater capacity to regenerate axons in peripheral nerves, but this cannot be enhanced further by activity-dependent treatments.
Introduction
Every year in the United States, there are more than 200 000 new cases of peripheral nerve injury (PNI). Despite the ability of axons in peripheral nerves to spontaneously regenerate, recovery is slow, and over 90% of adults who sustain a PNI never regain full motor function.1,2 Currently, there are no commonly used nonsurgical treatments for PNI. 3
Experimental treatments, such as exercise, electrical stimulation (ES), and optogenetic stimulation, have been shown to be effective in promoting elongation of regenerating axons, increasing recruitment of motor and sensory neurons into the regeneration process, and accelerating restoration of muscle responses to nerve stimulation.4-12 The success of these treatments requires increased activity of the participating neurons, 13 and thus they are collectively considered activity-dependent treatments. 14
Neuronal brain-derived neurotrophic factor (BDNF) secretion and signaling is required for the enhancing effects of activity-dependent treatments.15-17 Enhancement of axon regeneration is abolished in animals in which either BDNF or its trkB receptor have been selectively knocked out of neurons, indicating that neuronal BDNF as well as trkB activation are required for the enhancing effects of ES and exercise.15-18 This BDNF-dependence could impose a barrier to translation of activity-dependent treatments. About 30% of Americans have a single nucleotide polymorphism (SNP) in the bdnf gene resulting in replacement of valine by methionine at the 66th residue in the BDNF protein (Val66Met, see Figure 1A).19,20 BDNF is secreted through both a constitutive pathway and a regulated pathway. 21 Cells expressing the Met allele have deficits in regulated, calcium-dependent release of BDNF, but not constitutive release.19,22 This observation is mirrored by the finding that in Met-carriers, exercise-dependent increases in plasma BDNF are absent.23,24 This deficit in activity-dependent release of BDNF could inhibit the effectiveness of activity-dependent treatments for enhancing peripheral nerve regeneration.
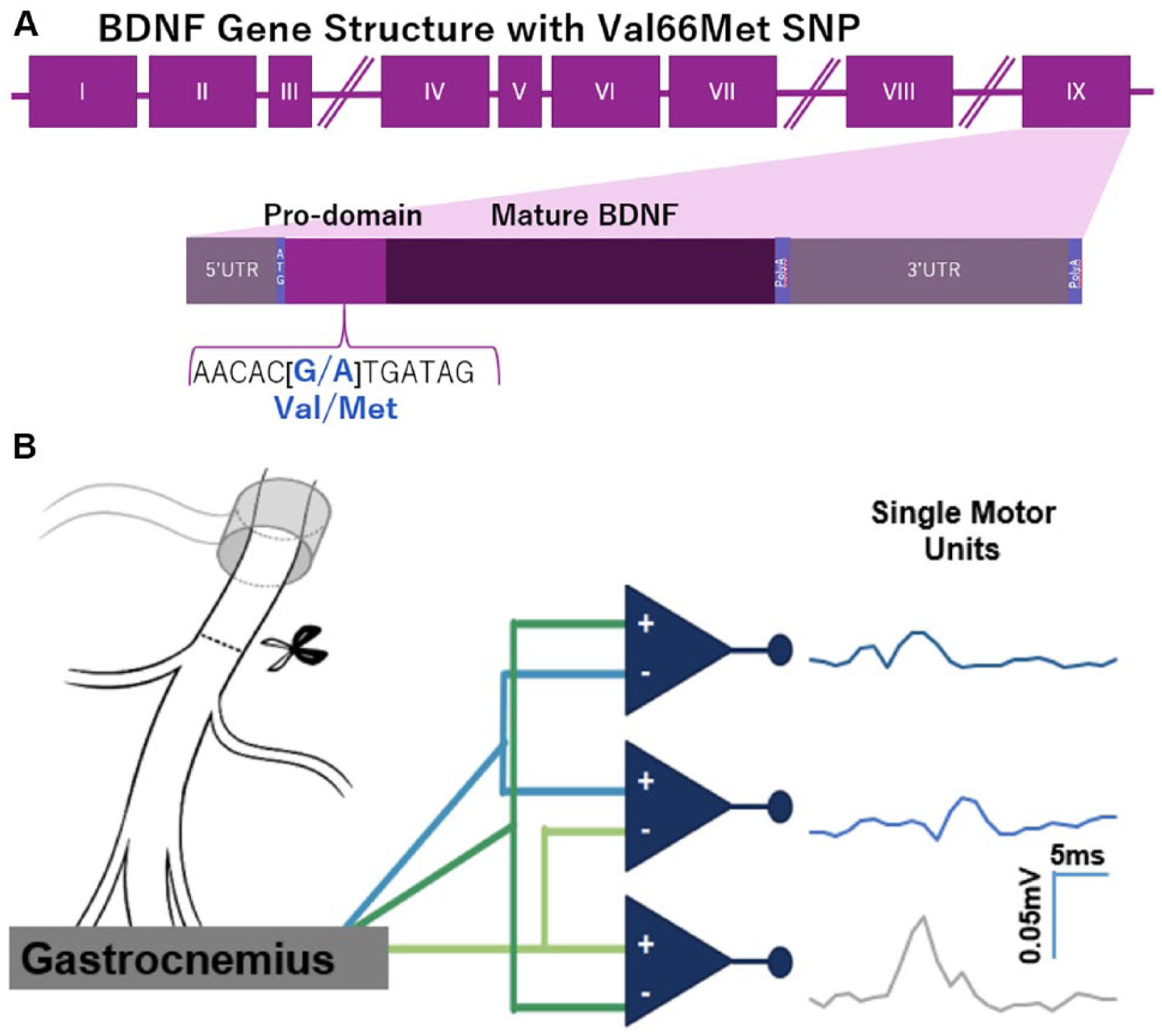
(A) Structure of BDNF gene and location of Val66Met SNP in the coding exon IX. The G to A substitution in the prodomain results in a valine to methionine substitution and a decreased Ca2+- dependent release of BDNF. (B) Recording single motor unit (SMU) potentials using a tripolar electrode. A stimulating cuff is placed around the sciatic nerve. A tripolar electrode is inserted into the lateral gastrocnemius muscle, and amplitudes of SMU potentials are recorded in three channels.
We tested the efficacy of 2 activity-based experimental therapies, treadmill training and optogenetic stimulation, in enhancing nerve regeneration after injury in mice heterozygous and homozygous for the Val66Met polymorphism.
Methods
Animals and Surgical Treatments
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Emory University and conformed to the Guidelines for the Use of Animals in Research of the Society for Neuroscience. Transgenic C57BL/6J mice heterozygous (V/M) and homozygous (M/M) for the met allele of the Val66Met polymorphism and their wild type littermates (V/V) were bred and maintained at the Division of Animal Resources at Emory University. Founders for these mice were a generous gift from Doctor Frances Lee. 25 Mice were group housed with ad libitum access to food and water. Both males and females were used in control experiments. Only females were used for exercise experiments.
Adult mice (over 2 months of age) were anesthetized with 2% isoflurane and treated with Meloxicam analgesic (2 mg/kg). The right sciatic nerve was exposed in the mid-thigh, placed on a small rectangle of SILASTIC film (Dow Corning 501-1) and secured with fibrin glue: a mixture of fibrinogen and thrombin (1:2, Sigma-Aldrich, St Louis, MO).26,27 The nerve was fully transected using sharp scissors three millimeters proximal to the sciatic branching. The aligned stumps on the mat were then secured with more fibrin glue. The left side of each mouse served as an unoperated control. After nerve repair, the surgical site was sutured closed, and the animal was allowed to recover from anesthesia before returning to its cage.
Treadmill Training
Female mice were exercised using an interval training protocol previously shown to be effective in enhancing axon regeneration. 28 Three days after transection surgery, mice began exercising. Animals performed 4 bouts of 2 minutes of high-intensity running (20 m/min), each separated by 5 minutes of rest. This training protocol was repeated 5 days per week for 2 weeks during their light cycle.
Motor Unit Number Estimation
Four weeks postinjury, the extent of muscle reinnervation was investigated using motor unit number estimation (MUNE). MUNE allows for the estimation of the number of functional motor units, in this case the number of motoneurons reinnervating the lateral gastrocnemius.29-31 Briefly, a stimulating cuff electrode was placed around the sciatic nerve proximal to the injury site, and 3 monopolar fine wire electromyography EMG electrodes (California Fine Wire Company, Grover Beach, CA) were implanted together in the lateral gastrocnemius muscle using a 25G hypodermic needle. The tips of the wires were hooked such that they were each 1 mm apart from each other. The wires were attached to 3 separate recording amplifiers, as shown in Figure 1B, to enable detection of distinct signals within the muscle. Electrical stimulation of the nerve (0.1-ms pulses applied every 2 seconds) was applied to evoke 3 compound muscle action potentials (CMAPs), which were recorded using custom LabVIEW software (National Instruments, Austin, TX). We have adapted the multiple point stimulation technique used in humans, in which small electrical currents were passed through the stimulating cuff and increased in amplitude until an all-or-nothing EMG response was noted.32,33 This lowest-threshold response on each channel was assumed to be produced by activation of a single motor unit (SMU). To be counted as an SMU, the response needed to be biphasic or triphasic and to occur more than once while stimulating voltage was held steady at the threshold needed for activation. The average rectified voltage within an empirically determined time window for each SMU potential was measured. 34 The stimulating cuff was moved to stimulate different points along the nerve until at least nine SMU potentials could be recorded. The stimulus intensity was then increased until a maximal CMAP was evoked. The amplitude of this potential was measured as above. It was assumed that this maximum CMAP was produced by the near synchronous activation of all of the motoneurons innervating the muscle. The average maximal CMAP amplitude for each animal was divided by the average of the SMU amplitudes to estimate the number of functional motor units in the reinnervated muscle. 35
Retrograde Labeling of Motoneurons
To determine the number of motoneurons that had successfully regenerated into the lateral gastrocnemius four weeks after injury, immediately following electrophysiological recordings, the muscles of both the intact and injured sides were exposed and each injected with 1 µL of a 1% solution of cholera toxin B (CTB) conjugated to a fluorescent label (Alexafluor 555; Life Technologies, Grand Island, NY, catalog number C-34776) as previously described. 12 Three days later, mice were perfused with aldehyde fixatives and lumbar spinal cords were harvested. Counts of labeled motoneurons were made from serial cryostat sections through the spinal cords as described in more detail elsewhere.12,36
Motor Endplate Reinnveration
The medial and lateral gastrocnemius muscles from both the intact and injured side of mice were harvested, cryoprotected in 20% sucrose, sectioned longitudinally (20 µm), and mounted onto slides. Sections underwent antigen retrieval in boiling 10 mM sodium citrate buffer (pH 8.5). Immediately following, sections were washed with 0.1 M PBS (phosphate buffered saline), blocked with 0.3% Triton in 0.1 M PBS and 10% natural goat serum for 1 hour at room temperature, and incubated at 4°C overnight (14-16 hours) with antibody against the vesicular acetylcholine transporter (VAChT, 1:500, Synaptic Systems, Gӧttingen, Germany) to label motoneuron synaptic terminals. Sections were washed in 0.1 M PBS and incubated for 2 hours at room temperature with secondary antibody, goat anti-guinea pig conjugated to Alexafluor 647 (1:200 Thermo Fisher Scientific, Waltham, MA) and α-bungarotoxin conjugated to Alexafluor 555 (αBT, 1:500, Sigma-Aldrich, Darmstadt, Germany) to label motor endplates. Glass coverslips were mounted using Entellan (Millipore, Darmstadt, Germany). For each muscle, 50 motor endplates were imaged and scored as reinnervated only if VAChT immunofluorescence completely filled the endplate (see Figure 4A).
Dorsal Root Ganglion Cell Culture
The V/M mice were bred with mice expressing the light–sensitive cation channel, channelrhodopsin, under the control of the Thy1 promoter (Thy1ChR2, https://www.jax.org/strain/007612). In these mice, a subset of deep root ganglion (DRG) neurons express ChR2.
11
In adult mice of both sexes (older than 2 months) euthanized with an overdose of isoflurane, entire vertebral columns were removed and DRGs were extracted and placed in cold Hank’s Balanced Salt Solution (HBSS, Corning, Corning, NY). After incubation in dispase (2.5 u/mL Sigma-Aldrich) and collagenase (200 u/mL Worthington Biochemical, Lakewood, NJ) in a 37°C bead bath for 45 minutes with gentle agitation applied every 15 minutes, DRGs were then treated in 37°C DNase (Worthington Biochemical) for 2.5 minutes before addition of room temperature HBSS. Cells were triturated using a fire-polished glass pipette and centrifuged for 3 minutes at 3000 rpm. The HBSS was removed and cells were resuspended in neurobasal medium A (NB-A, Invitrogen, Carlsbad, CA) containing 2% B-27 (Invitrogen), 1% penicillin/streptomycin (Lonza Biowhittaker), and 1% Glutamax (Invitrogen). Cells were seeded at a density of 1000 cells/well in 48-well Opticlear plates (Axion Biosystems, Atlanta, GA) coated in laminin (0.2 mg/mL, Thermo Fisher Scientific, Waltham, MA) and poly-
Twenty-four hours after plating, the NB-A solution was replaced. Plates were removed from the incubator and placed on an Axion Biosystems plate holder on top of a 37°C warming plate with 5% CO2 supplied. Cells were then stimulated using the Lumos System (Axion Biosystems) under control of their AxIS software (Axion Biosystems). Cells were exposed to 475 nm light at an intensity of 0.59 mW/mm2 at a rate of 20 Hz for 1 hour for a total of 72 000 pulses. 10 Control stimulation was the same regimen, but with light of an inappropriate wavelength (655 nm). Unstimulated cells received no light input. Media was collected immediately after stimulation and stored at −80°C for analysis of BDNF protein, as a significant increase in BDNF protein was found at that time in the media from DRG explants from Thy1ChR2 animals. 10 Analysis was performed with help from the Emory Multiplexed Immunoassay Core. Undiluted samples were analyzed in duplicate using the BDNF U Plex plates (Meso Scale Discovery, Rockville, MD). For protein analysis, one Thy1ChR2 V/M animal was excluded as an outlier as identified in the Grubb’s outlier test.
Seventy-two hours after plating, cells were fixed with 4°C periodate-lysate-paraformaldehyde fixative. Fixed cells were incubated with anti-tubulin β-3 antibody overnight (14-16 hours, Biolegend, San Diego, CA), followed by application of a secondary, goat anti-mouse antibody conjugated to Alexafluor 555 (Invitrogen) and DAPI (Invitrogen). Cells were imaged with a 20× objective on an epifluorescence microscope (Ti Eclipse; Nikon) equipped with a cooled charge-coupled device (CCD) camera (HQ2; Photometrics) using Nis Elements Imaging Software (Nikon). The cell soma area and longest processes from each cell were measured from these images using the Fiji software package (ImageJ). At least 30 cells per treatment were counted for each animal.
Experimental Design and Statistics
All results were scored while blinded to treatment and genotype. Data analyses were performed in Statistica 64, except for 95% confidence intervals (CI) and Mann-Whitney U tests, which were performed in Microsoft Excel. Power analyses were performed a priori (α = 0.05, power = 0.8) using data from previous studies to select adequate sample sizes for all outcome measures except for MUNE, which had not previously been performed in mice. For MUNE, power analysis was performed using preliminary data. To test the assumption of homoscedasticity required for analysis of variance (ANOVA), Levene’s test was used. Unless otherwise specified, one-way ANOVA was performed with Fisher’s LSD (least significant difference) post hoc test. For samples with unequal variance, Kruskal-Wallis ANOVA was performed. All results of statistical tests are reported in APA format.
Results
Functional Recovery Is Enhanced in Untreated Met-Carriers
Single motor unit potentials were recorded from intact mice and 4 weeks after injury with and without treadmill training (TT). Animals in 3 genotypes were studied: V/V, V/M, and M/M. The distributions of the amplitudes of these potentials are shown for the 3 genotypes in Figure 2A. As previously reported in both humans and rodents,37-39 in untreated V/V mice, the sizes of SMU potentials 4 weeks after injury were greater than those found in intact animals of the same genotype (Mann-Whitney test, U = 76, P = .009, Figure 2A, top panel). No significant difference in SMU amplitude relative to intact mice was found in animals of this genotype treated with TT. In V/M or M/M mice, no significant differences in the amplitudes of SMU potentials were found regardless of treatment (Figure 2A, bottom 2 panels).
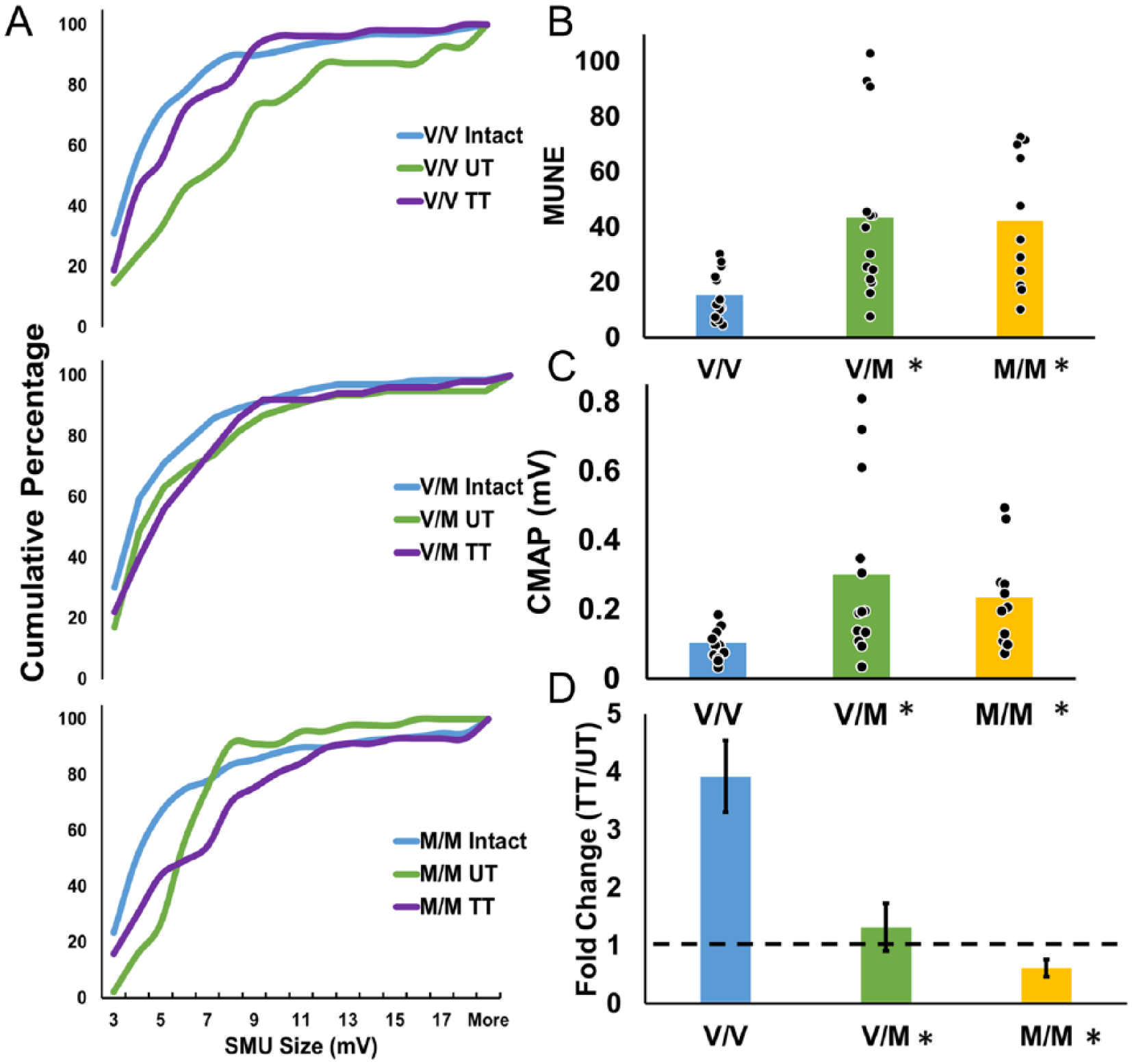
Treadmill training (TT) enhances functional recovery in V/V but not Met-carriers. (A) The distributions of single motor unit potential (SMU) amplitudes are shown for the different groups of mice studied as cumulative histograms. In V/V mice, sciatic nerve transection resulted in larger amplitude potentials 4 weeks after injury (top panel), and TT did not. In Met-carriers, SMU potential amplitude was not significantly different, regardless of injury or treatment (bottom 2 panels). (B) Motor unit number estimation (MUNE) is shown four weeks after injury in untreated mice of the three genotypes. Data from individual animals are solid symbols overlaid on the bars. Bars indicate mean value. Asterisks denote P < .05 with regard to V/V mice. (C) Maximum CMAP potential is shown 4 weeks after injury in untreated mice of the 3 genotypes studied. Data from individual animals are solid symbols overlaid on the bars. Bars indicate mean value. Asterisks denote P < .05 with regard to V/V mice. (D) Fold change in MUNE with TT is represented for the 3 genotypes. Data represented as mean (+SEM). Asterisks denote P < .05 with regard to V/V mice.
We used MUNE 35 to estimate the number of functional motor units in each muscle studied prior to and 4 weeks after sciatic nerve transection and repair. In intact animals, MUNE did not differ between the 3 genotypes, one-way ANOVA, F(2, 64) = 0.15, P = nonsignificant. Previous studies have not reported the effect of the met allele in females as well as males. Therefore, we tested the effect of sex and genotype on MUNE 4 weeks after transection and repair (n = 5-8). There was a significant effect of genotype, F(2, 32) = 6.0, P = .006, but not sex, so data from the UT males and females were pooled based on genotype. Once pooled, the Levene’s test for homogeneity of variance was significant (P = .0089), indicating nonhomoscedastic variance, so nonparametric Kruskal-Wallis ANOVA was used to analyze MUNE in untreated animals. A significant effect of genotype on MUNE, H(2) = 11.56, P = .0031, was found, with a mean rank of 11.00 for V/V, 23.86 for V/M, and 24.00 for M/M. Both V/M and M/M had a significantly higher mean rank than V/V (P = .0080 and P = .013m respectively, Figure 2B). A similar effect of genotype was found for maximal CMAP amplitude, H(2, 34) = 10.53, P = .0052; mean rank of 11.15 for V/V, 23.39 for V/M, and 23.09 for M/M, (Figure 2C). Based on post hoc testing, the maximal CMAP in V/V animals (0.10 ± 0.014 mV) was significantly smaller than V/M animals (0.30 ± 0.070 mV, P = .012) and M/M animals (0.23 ± 0.042 mV, P = .021).
The effect of treadmill training on motor unit reinnervation differed across genotypes. In treadmill trained V/V animals, a nearly 4-fold increase in MUNE was found relative to untreated controls (95% CI [2.72, 5.11] Figure 2C). In contrast, the fold change in treadmill trained V/M animals was not different from untreated (1.31-fold change, 95% CI [0.59, 2.04]). Treadmill training may actually result in decrease in the amount of motor unit reinnervation in M/M animals (95% CI [0.32, 0.90]). A significant difference in response to treadmill training was found between genotypes, Kruskal-Wallis ANOVA, H(2) = 11.09, P = .0039, with a mean rank of 15.17 for V/V, 8.17 for V/M, and 5.17 for M/M. Ranks for V/V and M/M were significantly different from each other (P = .0035).
Motor Axon Regeneration Is Enhanced in Untreated but Not Treadmill-Trained Mice Heterozygous for BDNFmet
To assay the extent of successful motor axon regeneration, we injected a retrograde tracer bilaterally into the lateral gastrocnemius muscles and counted the number of labeled motoneurons in cryostat section of the lumbar spinal cord (Figure 3A). The ratio of counts of the injured side to the intact side of each mouse was used to evaluate the proportion of motoneurons reinnervating the muscle. Four weeks after peripheral nerve injury, fewer than half of injured motoneurons had regenerated into the lateral gastrocnemius muscle in untreated V/V mice (0.45 ± 0.077, Figure 3B) as has been shown previously. 12 Regeneration in untreated V/M mice was almost twice that of V/V (0.81 ± 0.068). Motoneuron regeneration in M/M mice was similar to V/V mice at 0.50 ± 0.16. The result of a one-way ANOVA comparing the 3 genotypes (V/V, V/M, and M/M) was significant, F(2, 12) = 6.31, P = .01. Based on post hoc paired testing, the injured/intact ratio in untreated V/M mice was significantly larger than that in either untreated V/V mice or M/M mice (P = .009 and P = .01, respectively, Figure 3B). There was no significant difference between untreated V/V and M/M mice (P = nonsignificant).
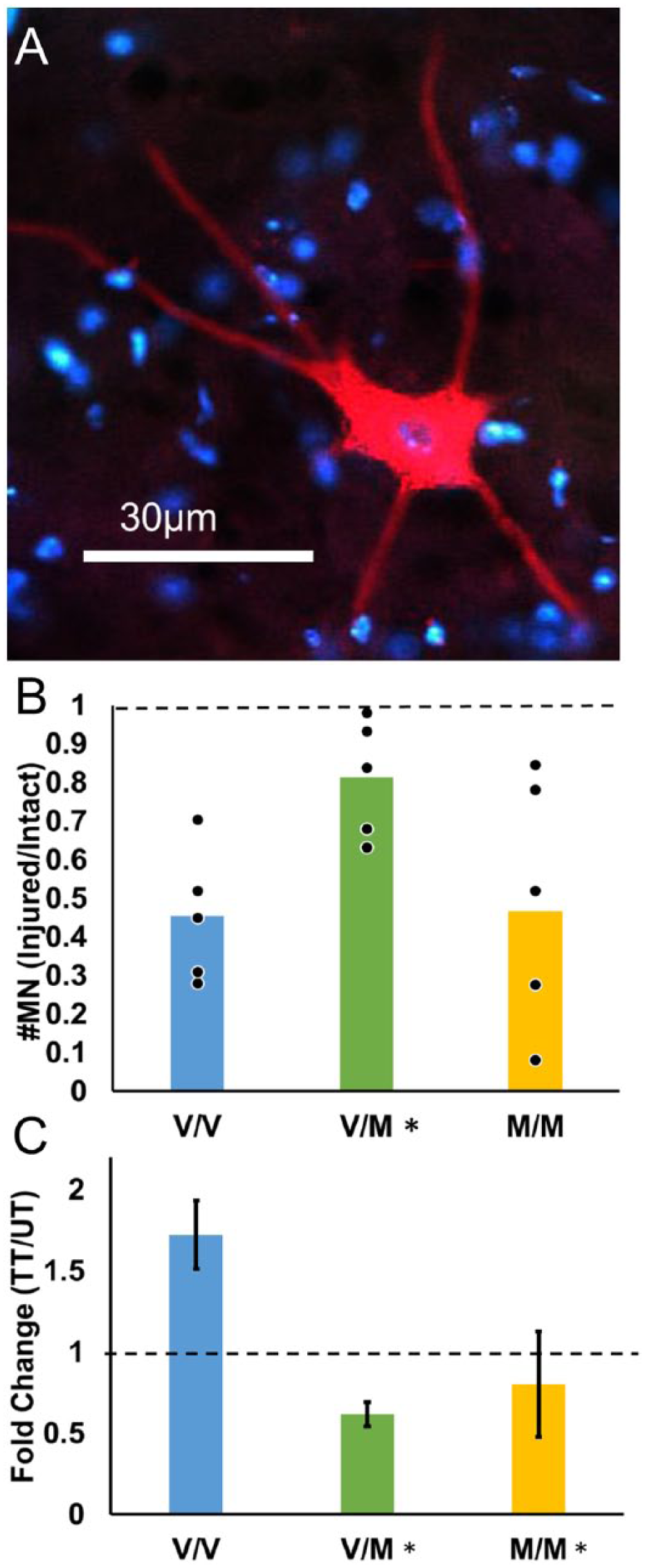
Motor axon regeneration is enhanced in untreated but not treadmill-trained (TT) mice heterozygous for BDNFmet. (A) A single motoneuron labeled by a retrograde tracer that had been injected into the lateral gastrocnemius muscle 4 weeks after sciatic nerve transection and repair. Motoneurons like this one were counted as labeled if the soma was labeled with dense granular fluorescence which extends into the primary dendrites and contains a dark region, indicating a nucleus. Red is CTB-555, blue is DAPI. (B) The ratio of the number of labeled motoneurons (injured/intact) is shown. Data from individual animals are solid symbols overlaid on the bars. Bars indicate mean value. Asterisks denote P < .05 with regard to V/V mice. (C) Mean (±SEM) fold change in the number of retrogradely labeled motoneurons in TT animals relative to untrained controls (UT) are shown for the 3 genotypes studied (V/V, N = 5; V/M N = 6, M/M, N = 5). Data represented as mean (±SEM). Asterisks denote P < .05 with regard to V/V animals.
To analyze the effect of genotype on response to treadmill training, the fold change (TT/UT) in number of retrogradely labeled motoneurons was calculated. Treadmill training increased the number of labeled motoneurons in V/V mice 1.72-fold (95% CI [1.31, 2.13]). In contrast, treadmill training may have been detrimental to motoneuron regeneration in V/M mice (0.62-fold change, 95% CI [0.47, 0.76]). Treadmill training had no effect on motoneuron regeneration in M/M mice (0.93-fold change, 95% CI [0.20, 1.66]). The result of a one-way ANOVA was significant, F(2, 13) = 7.7, P = .006, Figure 3C. Using post hoc paired testing, significant differences in the fold-change of the proportion of labeled motoneurons were found between V/V and V/M mice (P = .002) and V/V and M/M mice (P = .01). No difference was found between treadmill trained V/M and M/M mice (P = nonsignificant).
Muscle Fiber Reinnervation Is Enhanced in Untreated but Not Treadmill-Trained Mice Heterozygous for BDNFmet
Muscle fiber reinnervation was analyzed by determining the proportion of alpha-bungarotoxin-positive motor endplates where VAChT, a marker of cholinergic motoneuron terminals, was expressed. Only motor endplates with complete coverage by VAChT immunofluorescent structures were counted (Figure 4A). With no treatment, 18.6% ± 8.06% of motor endplates in V/V animals were occupied by motoneuron terminals 4 weeks after sciatic nerve transection and repair (Figure 4B). In untreated V/M mice, 46.3% ± 9.84% of motor endplates were occupied by motoneuron terminals, and in M/M mice, 22% ± 2.37% of motor endplates were occupied. The results of a one-way ANOVA of the 3 genotypes were significant, F(2, 15) = 4.11, P = .038). Post hoc testing revealed a significantly greater percentage of VAChT-covered motor endplates in untreated V/M mice than in untreated V/V mice (P = .016) and M/M mice (P = .046) (Figure 4B). In V/V animals, treadmill training resulted in a 2.6-fold increase in percent of VAChT positive motor endplates (95% CI [1.71, 3.57], Figure 4C). In contrast, treadmill training had no effect on percentage of VAChT positive motor endplates in V/M mice (95% CI [−0.40, 1.45]) or M/M mice (95% CI [0.53, 2.39]). Levene’s test of variance was significant (P = .0077). There was a significant in response to treadmill training between genotypes, H(2) = 7.04, P = .030, with a mean rank of 13.50 for V/V, 5.33 for V/M, and 9.67 for M/M. Post hoc testing revealed a significant difference between V/V and V/M mice (P = .024).
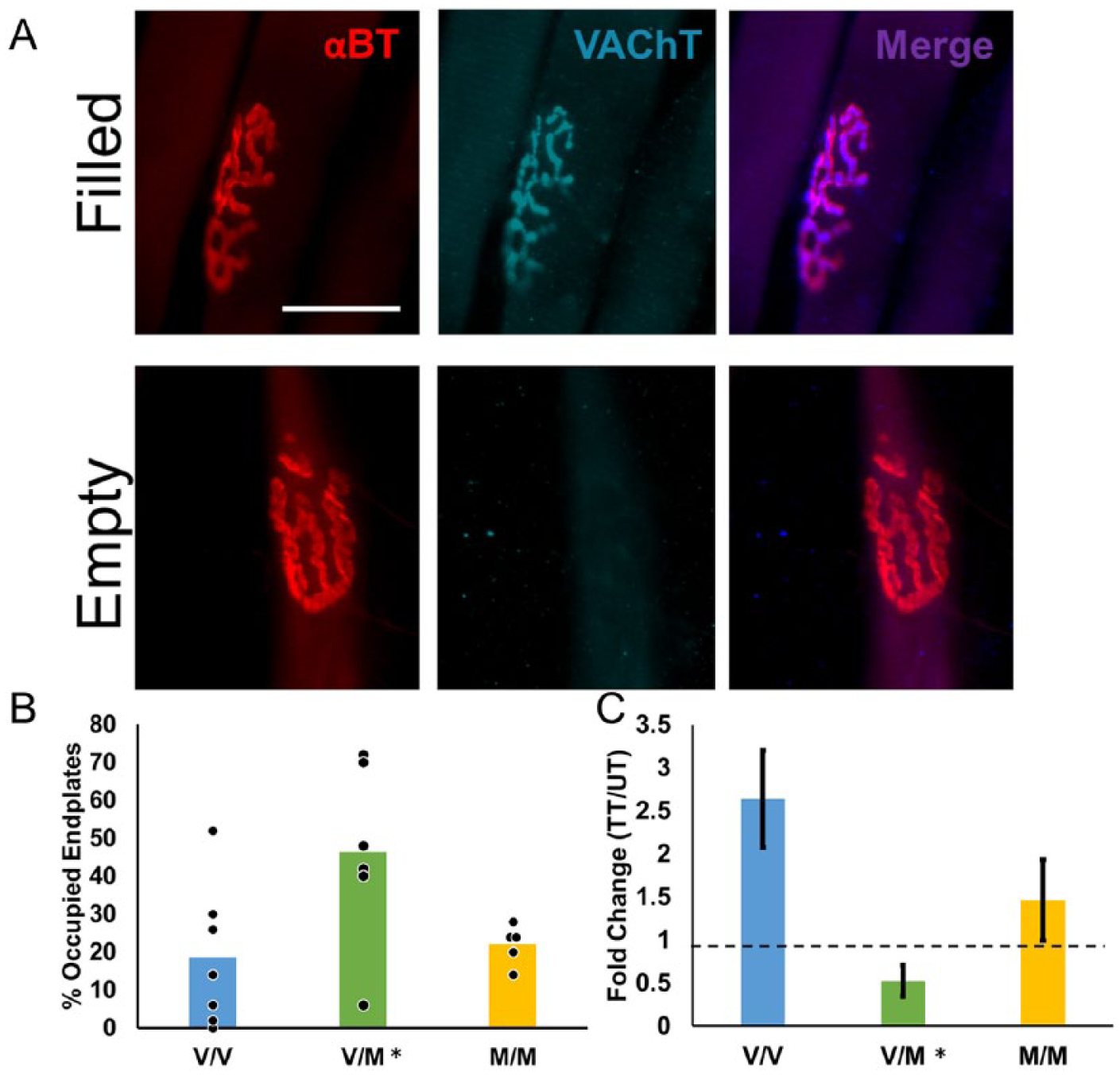
Muscle fiber reinnervation is enhanced in untreated but not treadmill-trained (TT) mice heterozygous for BDNFmet. (A) Using the binding of fluorescent alpha-bungarotoxin (αBT), 2 motor endplates are shown (top and bottom left panels). Immunoreactivity to VAChT marks motoneuron terminals (top and bottom middle panels). Motor endplates were counted as filled if the VAChT completely covered the motor endplate (top panels). Fifty motor endplates were scored for each animal. Scale bar = 25 µm. (B) A higher percentage of motor endplates were scored as VAChT+ in muscles harvested from untreated V/M mice than V/V or M/M mice. Data from individual animals are solid symbols overlaid on the bars. Bars indicate mean value. Asterisks denote P < .05 with regard to V/V mice. (C) Effect of TT on motor endplate reoccupation. Mean (±SEM) fold change in TT animals relative to untrained controls (UT) are shown for the 3 genotypes studied (V/V, N = 6; V/M N = 6, M/M, N = 6). Asterisks denote P < .05 with regard to V/V mice.
Neurite Outgrowth Is Enhanced in V/M and M/M Neurons Without Optogenetic Stimulation
Adult DRG neurons derived from mice expressing the light-sensitive cation channel, channelrhodopsin, were cultured for 72 hours. Wild type (WT) neurons were also cultured as light-insensitive controls. To evaluate the effects of activity and genotype on neurite outgrowth, the longest neurite length for each neuron was measured. Figure 5A is a representative photo of Thy1ChR2 V/V DRG neurons. Seventy-two hours after plating, the average longest neurite length for untreated Thy1ChR2 V/V neurons was 235.22 ± 4.6 µm (Figure 5B). Thy1ChR2 V/M and Thy1ChR2 M/M neurons grew longer neurites, with averages of 325.10 ± 8.8 and 357.22 ± 22.1 µm, respectively. The result of one-way ANOVA was significant for an effect of genotype, F(3, 12) = 15.84, P < .001. Average longest neurite length from Thy1ChR2 V/V neurons was significantly shorter than average neurite length from Thy1ChR2 V/M neurons (P = .013) and Thy1ChR2 M/M neurons (P < .001), but not WT neurons (P = nonsignificant). Thy1ChR2 V/M and Thy1ChR2 M/M neurons did not differ in longest neurite length (P = nonsignificant).
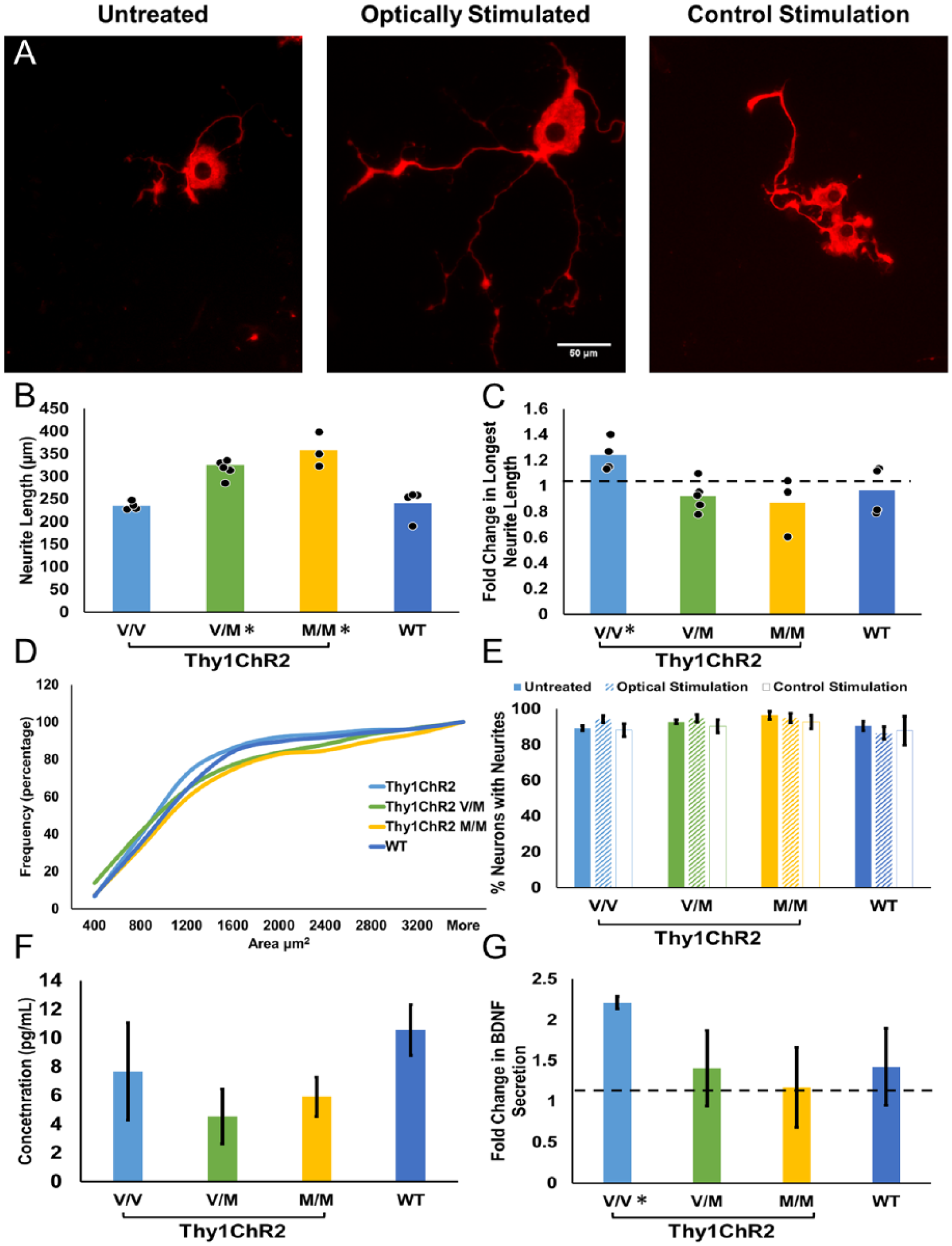
(A) Representative images of cultured V/V DRG neurons immunofluorescent for beta tubulin III. Cells were cultured from adult mice both male and female. (B) The mean longest neurite length is shown for untreated cultures of the 4 genotypes studied. Data from individual cultures are shown as solid symbols overlaid on the bars. Asterisks denote P < .05 with regard to Thy1ChR2 V/V mice. (C) Fold-change in longest neurite length after 1-hour light stimulation is shown for the 4 genotypes studied. Data from individual cultures are shown as solid symbols overlaid on the bars. Asterisk denotes significant difference from 1. (D) A cumulative histogram of the soma size distributions of cultured DRG neurons for the 4 genotypes. There was no significant bias in the sizes of the neurons studied in any of the groups. (E) The percent of neurons that grew neurites is shown for the 4 genotypes. Cells were untreated, stimulated optically with 472 nm light, or stimulated with control stimulation of light of an inappropriate wavelength. Data are represented as mean (±SEM). (F) Media was collected for protein analysis of basal levels of secreted BDNF protein. Data are represented as mean (±SEM). (G) Immediately after light stimulation, media was collected for protein analysis of BDNF secretion. Fold-change of BDNF secretion is represented. Data are expressed as mean (±SEM). Asterisks denote P < .05 with regard to 1.
Optogenetic Stimulation Enhances Neurite Elongation in V/V Neurons In Vitro
To measure neurite outgrowth in response to activity-dependent treatment, cells were exposed to 1 hour of 20 Hz light stimulation at 24 hours in culture and were fixed and stained 48 hours later. Longest neurite length was measured, and fold-change (optically stimulated/untreated) was calculated. A 1.2-fold change in neurite length was recorded in Thy1ChR2 V/V cells stimulated with light (95% CI [1.12, 1.36], Figure 5C). In contrast, no response to light stimulation was found in the other 3 genotypes (Thy1ChR2 V/M 0.92-fold change, 95% CI [0.82, 1.03]; Thy1ChR2 M/M 0.87-fold change, 95% CI [0.61, 1.13]; WT 0.96-fold change, 95% CI [0.78, 1.15]). The result of a one-way ANOVA was significant, F(3, 12) = 4.03, P = .034. Post hoc testing revealed Thy1ChR2 V/V fold change was significantly different from Thy1ChR2 V/M fold change (P = .013), ThyChR2 M/M fold change (P = .011), and WT fold change (P = .034). As a control, cells were also stimulated with an inappropriate wavelength of light (655 nm). Neurite length did not differ with this control stimulation in any genotype, and there was no difference between any of the genotypes (P = nonsignificant).
Differences in longest neurite length could be due to neurons growing longer neurites or differences in the proportion of cells that grew any neurites. To test this, we analyzed the proportion of cells with neurites for each genotype and treatment group, and found no effect of genotype, F(2, 18) =1.79, P = nonsignificant; treatment, F(1, 18) = 1.27, P = nonsignificant; or an interaction of genotype ∗ treatment, F(2, 18) = 1.20, P = nonsignificant (Figure 5E). There also was no significant difference between genotypes or treatments in the soma sizes of the cultured neurons studied (see Figure 5D).
There Is No Difference in Basal Release of BDNF Between Genotypes
Immediately after light stimulation, media was collected from the DRG culture for analysis of secreted BDNF protein (Figure 5F). The result of a one-way ANOVA revealed no significant effect of genotype on BDNF secretion in unstimulated DRG neurons, F(3, 11) = 1.10, P = nonsignificant. Optogenetic stimulation increased BDNF concentration 2-fold in media collected from Thy1ChR2 V/V cells (Figure 5G, 2.21, 95% CI [2.059, 2.306]). There was no difference in BDNF concentration in media collected from stimulated Thy1ChR2 V/M neurons (1.40, 95% CI [0.50, 2.31]), Thy1ChR2 M/M neurons (1.17, 95% CI [0.216, 2.13]), or WT neurons (1.42, 95% CI [0.36, 2.49]). One-way ANOVA revealed no significant effect of genotype on fold-change of BDNF concentration, F(3, 10) = 1.30, P = nonsignificant.
Discussion
Activity-dependent treatments have proven useful in enhancing axon regeneration after peripheral nerve injury. Their effectiveness requires BDNF-trkB signaling in the neurons whose axons are regenerating. We used neuroanatomical and electrophysiological methods to analyze the ability of one such treatment, treadmill training, to enhance axon regeneration in mice carrying the Met allele of the Val66Met SNP, which have deficient activity-dependent release of BDNF. Here, we present 2 main findings. First, without treatment, axon regeneration in Met-carriers is enhanced in vivo and neurite outgrowth is longer in vitro when compared with non-Met carriers. Second, treadmill training, which is a powerful activity-dependent promoter of axon regeneration, did not enhance regeneration in Met-carriers in vivo, and optogenetic stimulation did not enhance neurite outgrowth in vitro.
Without any treatment, motor axon regeneration in Met-carriers was enhanced significantly compared with non-Met-carriers. Our results from MUNE, retrograde labeling, and analysis of motor endplate reinnervation support the conclusion that axons of more motoneurons effectively regenerated and reinnervated a muscle target in untreated V/M and M/M mice. The simplest interpretation of these findings is that the process of regeneration/reinnervation is accelerated in these animals. Our finding that neurites from cultured DRG neurons expressing the Met allele were significantly longer than those derived from V/V mice is also interpreted as more rapid regeneration.
The enhanced regeneration of axons in the Met-carriers was an unexpected result. The valine to methionine replacement results in inefficient packaging of BDNF into calcium-sensitive vesicles, making activity-dependent release of BDNF deficient in V/M and M/M mice.19,22 These vesicles release BDNF in response to calcium influx, such as might occur in motoneurons during exercise. 21 In contrast, no difference in basal release of BDNF has been reported.25,40 Thus, we did not anticipate improved regeneration in our untreated V/M and M/M mice.
The mechanism behind the observed enhanced regeneration/neurite outgrowth in Met-carriers is currently unknown. One possibility could be greater constitutive release of BDNF resulting from the deficiency in regulated release described above. However, we found no difference in basal levels of BDNF secretion in our culture experiments, consistent with previous studies.25,40 Another possible explanation could be increased trkB expression in the regenerating the axons, resulting in more effective ligand-binding despite less available ligand. Higher levels of full length trkB and lower levels of the dominant-negative truncated form of trkB have been found in the dorsal hippocampus in M/M mice. 40 A similar elevation of trkB expression in DRG neurons and motoneurons in mice with the Met allele could be a part of a mechanism explaining the enhanced axon growth observed. Additionally, compensatory changes in other ligands for trkB, such as neurotrophin 4/5 (NT4/5), could also account for differences in axon regeneration. Neuronal release of NT4/5 contributes to axon regeneration after injury,41,42 though basal levels of neuronal NT 4/5 secretion have not been studied, nor has activity-dependent secretion. 21 These possible mechanisms should be explored further in future studies.
Carrying the Met allele of the BDNF Val66Met polymorphism has been reported as a risk factor for numerous diseases and disorders, including Alzheimer’s disease, obsessive compulsive disorder, anorexia nervosa, and bipolar disorder.43-47 Here, we find a possible beneficial result of this allele—better axon regeneration after injury. We are not the first to report a surprising benefit of carrying the Met allele. Met-carriers more effectively recover executive functioning after traumatic brain injury and have a lower risk of mortality.48-50 In stroke models, the Met allele was associated with enhanced motor performance after a transient middle cerebral artery occlusion. 51 These findings taken together could indicate that having this allele is not simply bad—indeed, it would be hard to explain the high prevalence of the allele in East Asian populations if the results were unequivocally negative. 20
Neither treadmill training nor optogenetic stimulation further enhanced regeneration in Met-carriers. This result was expected due to the BDNF-dependence of these treatments and the deficient activity-dependent BDNF-release found in Met-carriers.17,19,25 Treadmill training markedly improved MUNE, motoneuron labeling, and motor endplate occupation in V/V mice.
Conclusion
Clinical trials testing the efficacy of activity-dependent treatments for nerve damage have already begun.37,52 In these studies, brief electrical stimulation is applied after carpal tunnel release surgery, resulting in increased MUNE 6 to 8 months after surgery. While promising, the enhancing effects of electrical stimulation could be diminished by inclusion of Met-carriers, who, based on the results presented above, might be expected to have naturally better outcomes under control conditions and not to respond to this activity-dependent treatment. Without genotyping of patients, benefits of activity-dependent treatments for non-met-carriers could be masked. More importantly, as activity-based treatments increase in popularity to treat peripheral neuropathies, health care professionals need to account for patient genotype. For the substantial portion of the population carrying the Met allele, personalized medicine that does not rely on endogenous BDNF secretion may be necessary.
Footnotes
Acknowledgements
Special thanks to Axion Biosystems for the use of their Lumos multiwell light delivery device and AxIS software. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health. Many thanks to Dr Francis Lee for the gift of the Val66Met mouse. Many thanks also to Dr Gary Bassell and Phillip Price for assistance with cell culture.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Support provided by grant NS057190 to AWE from the USPHS. This study was supported in part by the Emory Multiplexed Immunoassay Core (EMIC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. Additional support was provided by the National Center for Georgia Clinical & Translational Science Alliance of the National Institutes of Health under Award Number UL1TR002378.