
Abstract
Background. Neutralization of central nervous system neurite growth inhibitory factors, for example, Nogo-A, is a promising approach to improving recovery following spinal cord injury (SCI). In animal SCI models, intrathecal delivery of anti-Nogo-A antibodies promoted regenerative neurite growth and functional recovery. Objective. This first-in-man study assessed the feasibility, safety, tolerability, pharmacokinetics, and preliminary efficacy of the human anti-Nogo-A antibody ATI355 following intrathecal administration in patients with acute, complete traumatic paraplegia and tetraplegia. Methods. Patients (N = 52) started treatment 4 to 60 days postinjury. Four consecutive dose-escalation cohorts received 5 to 30 mg/2.5 mL/day continuous intrathecal ATI355 infusion over 24 hours to 28 days. Following pharmacokinetic evaluation, 2 further cohorts received a bolus regimen (6 intrathecal injections of 22.5 and 45 mg/3 mL, respectively, over 4 weeks). Results. ATI355 was well tolerated up to 1-year follow-up. All patients experienced ≥1 adverse events (AEs). The 581 reported AEs were mostly mild and to be expected following acute SCI. Fifteen patients reported 16 serious AEs, none related to ATI355; one bacterial meningitis case was considered related to intrathecal administration. ATI355 serum levels showed dose-dependency, and intersubject cerebrospinal fluid levels were highly variable after infusion and bolus injection. In 1 paraplegic patient, motor scores improved by 8 points. In tetraplegic patients, mean total motor scores increased, with 3/19 gaining >10 points, and 1/19 27 points at Week 48. Conversion from complete to incomplete SCI occurred in 7/19 patients with tetraplegia. Conclusions. ATI335 was well tolerated in humans; efficacy trials using intrathecal antibody administration may be considered in acute SCI.
Introduction
Worldwide, the incidence of traumatic spinal cord injury (SCI) ranges from 3.6 to 195.4 patients per million, with most cases occurring in young adults.1,2 The psychosocial and physical consequences of SCI are devastating, with very few people with severe SCI recovering full neurological function.3-6 Specific treatments targeted to the injured spinal cord are limited. Methylprednisolone was a previously recommended treatment in acute SCI; however, because of limited clinical effectiveness and associated adverse events (AEs), updated guidelines either no longer support the use of methylprednisolone or suggest a low-dose protocol as a treatment option to be given within the first 8 hours of injury.7-13
Central nervous system (CNS) recovery following both SCI and brain trauma is, in part, limited by myelin-associated inhibitors of neurite growth, including Nogo-A, an oligodendrocyte membrane protein composed of multiple functional domains that interact with neuronal receptors.14,15 Nogo-A is one of the best-known inhibitors of neurite growth and plasticity in adult CNS, where it restricts long-distance axon growth and regeneration and stabilizes neuronal circuits. 15 Treatment with anti-Nogo-A antibodies neutralized the inhibitory activity on neurite growth of purified or recombinant Nogo-A, oligodendrocytes, and CNS myelin in vitro.14-18 In rodent and nonhuman primate models of partial and complete SCI, intrathecal (i.t.) delivery of anti-Nogo-A antibodies promoted axon growth, including collateral sprouting, and improved functional recovery.19-21
ATI355 is a recombinant human antibody (immunoglobulin G [IgG]4/κ class) directed against the human Nogo-A protein.22,23 ATI355’s mechanism of action includes steric hindrance of the Nogo-A inhibitory domains and antibody-induced downregulation of the Nogo-A protein.24,25
This first-in-man cohort study assessed the feasibility, acute safety and tolerability, pharmacokinetics (PK), and preliminary efficacy of ATI355 administered into the cerebrospinal fluid (CSF) compartment by continuous i.t. infusion or repeated i.t. bolus injections in patients with acute traumatic complete paraplegia and tetraplegia.
Methods
Experimental Design
This was a Phase I, open-label, multicenter cohort study of ATI355 administered by continuous i.t. infusion by external pump or repeated i.t. bolus injections in patients with acute traumatic paraplegia and tetraplegia (ClinicalTrials.gov identifier: NCT00406016; https://clinicaltrials.gov/ct2/show/NCT00406016).
A minimum of 40 patients were planned to be enrolled in 6 partially overlapping, sequential cohorts (Cohorts I, II, III, IV, IVb, and V; Figure 1); the relatively low enrolment was due to stringent safety criteria (see Supplementary Methods for rationale and details of cohort progression).
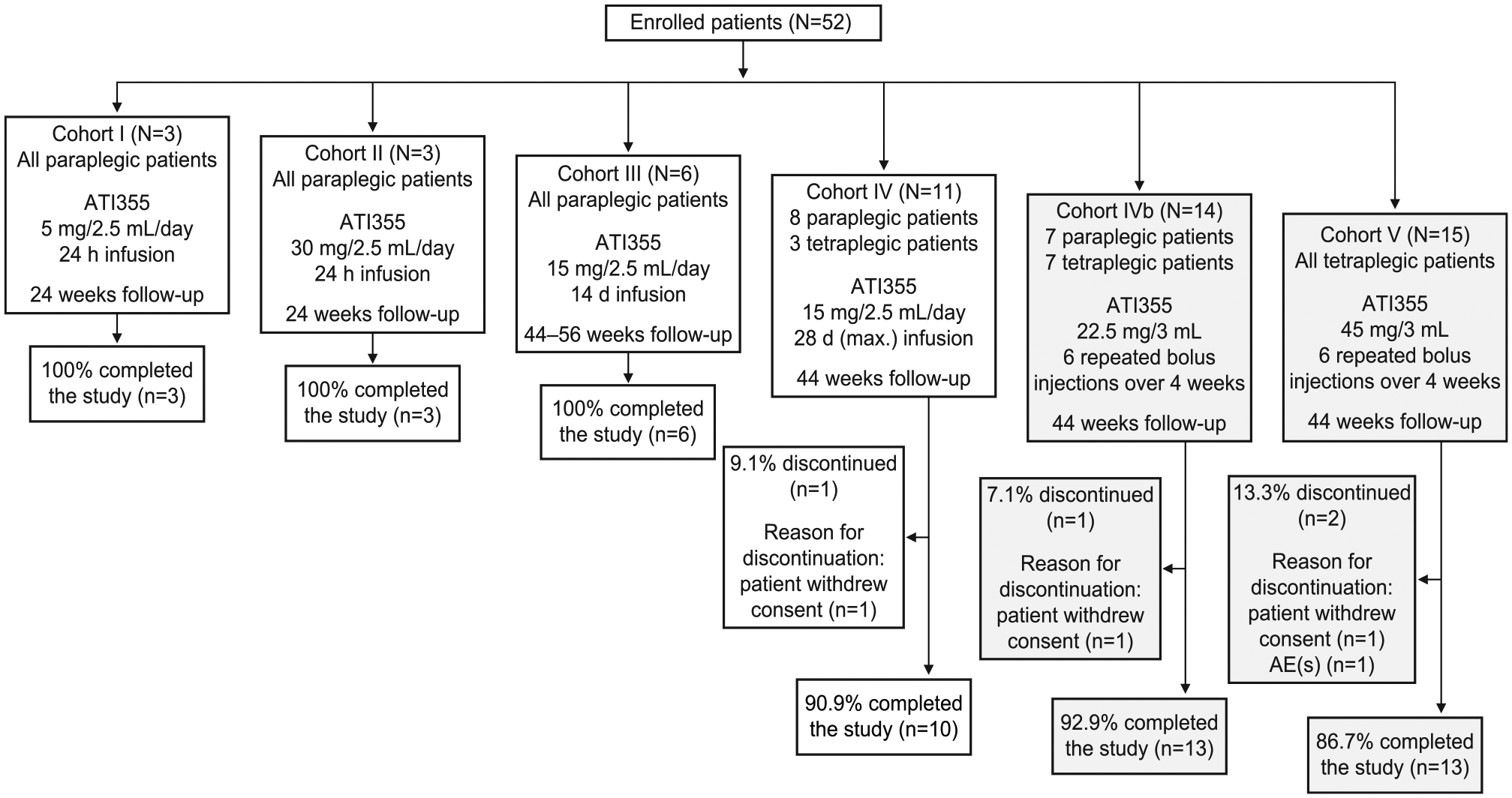
Disposition of patients.
Cohorts I to III assessed the feasibility of the i.t. administration route and the safety and tolerability of continuous infusion of ATI355 in patients with thoracic SCI. Cohort IV included paraplegic and tetraplegic patients who received ATI355 via i.t. infusion. Cohorts IVb (paraplegic and tetraplegic patients) and V (tetraplegic patients) investigated the safety and tolerability of repeated bolus injections of ATI355.
Patients were hospitalized for the treatment duration and ≥2 weeks postinfusion. Any patients not completing ATI355 treatment were requested to complete all subsequent safety visits and assessments.
The study protocol and amendments (see Supplementary Appendix for protocol amendments) were approved by the Ethik-Kommission der Medizinischen Fakultät Heidelberg, Heidelberg, Germany (#AFmu-485/2005), the Ethik-Kommission (KEK), SPUK Orthopaedie, Universität Zürich, Zürich, Switzerland (#27/2005), and the Office of Medical Bioethics, Cojoint Health Research Ethics Board, Faculty of Medicine, University of Calgary, Calgary AB, Canada (#E-20902). The study followed good clinical practice and was conducted in accordance with the Declaration of Helsinki. Informed written consent was obtained from all patients.
Patients
This study was conducted at SCI centers in Switzerland, Germany, and Canada. Adults 18 to 65 years of age with acute traumatic, complete SCI (American Spinal Injury Association [ASIA] Impairment Scale [AIS] A, according to the International Standards for Neurological Classification of SCI [ISNCSCI] 26 ) were eligible to participate. Patients in Cohorts I to III and the first 3 patients in Cohorts IV/IVb had paraplegia due to thoracic lesions (neurological level of injury T2 through T12) and received ATI355 4 to 14 days postinjury. In Cohorts IV/IVb, patients with thoracic or cervical lesions (neurological level of injury C5 through T12), who were not receiving ventilation, initiated ATI355 4 to 14 days postinjury. For tetraplegic patients requiring ventilation, treatment could be initiated up to 60 days postinjury (as soon as the patient was off ventilation). In Cohort V, tetraplegic patients with cervical lesions (neurological level of injury C5 through T1) showing some degree of spontaneous respiration (ie, not requiring or not completely dependent on ventilation) began ATI355 treatment 4 to 28 days postinjury.
Key exclusion criteria included complete anatomical transection confirmed by magnetic resonance imaging (MRI); trauma caused by ballistic or other injury that directly penetrated the spinal cord or MRI indicating complete obstruction of the i.t. space (see Supplementary Methods for a full list of inclusion and exclusion criteria).
Study Drug
Cohorts I to IV were treated on Day 1 with continuous infusion of ATI355; patients in Cohorts IVb and V received bolus injections of the study drug, receiving the first injection on Day 1 (see Figure 1 for dosage information and length of follow-up periods, and Supplementary Methods for the administration procedures).
Primary Objectives
To evaluate the feasibility, acute safety and tolerability, and serum PK and CSF concentrations of ATI355 administered by continuous i.t. infusion or repeated bolus injections in traumatic paraplegic and tetraplegic patients.
Secondary Objectives
To assess the immune response to ATI355 in patients with acute SCI and investigate an early potential efficacy signal (ISNCSCI including AIS, and motor and sensory scores) and/or electrophysiological changes (somatosensory-evoked potential [SSEP], motor evoked potentials [MEP], nerve conduction velocity [NCV]) in this patient population.
Safety Assessments
Safety assessments included physical examinations, electrocardiogram (ECG), vital signs and standard clinical laboratory evaluations (hematology, blood chemistry, and urinalysis), and monitoring of AEs and serious AEs (SAEs).
PK and Immunogenicity Assessments
Serum and CSF samples were collected for measurement of PK and immunogenicity. Timings of serum and CSF sampling for both measurements are shown in Supplementary Tables 1 to 4. Details of how ATI355 concentrations were measured are included in the Supplementary Methods.
Neurological Assessment
The potential impact of ATI355 on neurological recovery was evaluated using the ISNCSCI motor and sensory scores and AIS grade (see Supplementary Methods). Timings of neurological assessments made at screening, baseline (Day −7 to Day −1), and during treatment are shown in Supplementary Tables 1 to 4.
Electrophysiological Tests
Electrophysiological tests (SSEP, MEP, and NCV) were conducted at baseline and follow-up27,28 to assess patients’ neurological conductivity (see Supplementary Methods).
Sample Size
As the first-in-man study, the numbers of patients were limited to 3, 3, 6, 10, 12, and 16 (maximum) in Cohorts I to V, respectively. For Cohort V, a minimum of 6 to 8 patients were needed to compare with Cohort IVb data. Cohort size was determined by safety data from the previous cohort (see Supplementary Methods).
Statistical Analysis
All patients who received i.t. catheterization or repeated bolus injections were included in the data analysis. In the analysis of primary variables, vital signs, ECG, and laboratory data were listed by cohort, patient, or visit; summary statistics were provided. AE data were tabulated by preferred term for each cohort. Serum and CSF PK data were summarized using descriptive statistics. All patients with quantifiable/complete PK data were included in the PK analysis. In the analysis of secondary variables, neurological recovery/pharmacodynamics data were listed by cohort and patient; summary statistics for the change from baseline to the study endpoint on ISNCSCI motor and sensory scores were provided.
Results
Patients
Fifty-two eligible patients were enrolled in the study, of whom, 48 (92%) completed the study. One patient each in Cohorts IV, IVb, and V withdrew their consent, and 1 patient in Cohort V discontinued from the study due to an AE (lung embolism; Figure 1).
Mean ages of patients were similar in Cohorts II to IVb (range 32.3-35.9 years); patients tended to be older in Cohort I (mean [standard deviation], 45.3 [17.6] years) and younger in Cohort V (28.3 [13.3] years) than the other cohorts (Table 1). Most patients were male and Caucasian. The mean height, weight, and body mass index of patients were similar across all cohorts (Table 1). The mean (range) time from injury to receiving ATI355 treatment was 11.9 (6, 15) days in Cohorts I to III, 17.4 (3, 53) days in Cohorts IV/IVb, and 22.3 (14, 27) days in Cohort V.
Patient Demographics at Baseline for Each Cohort a
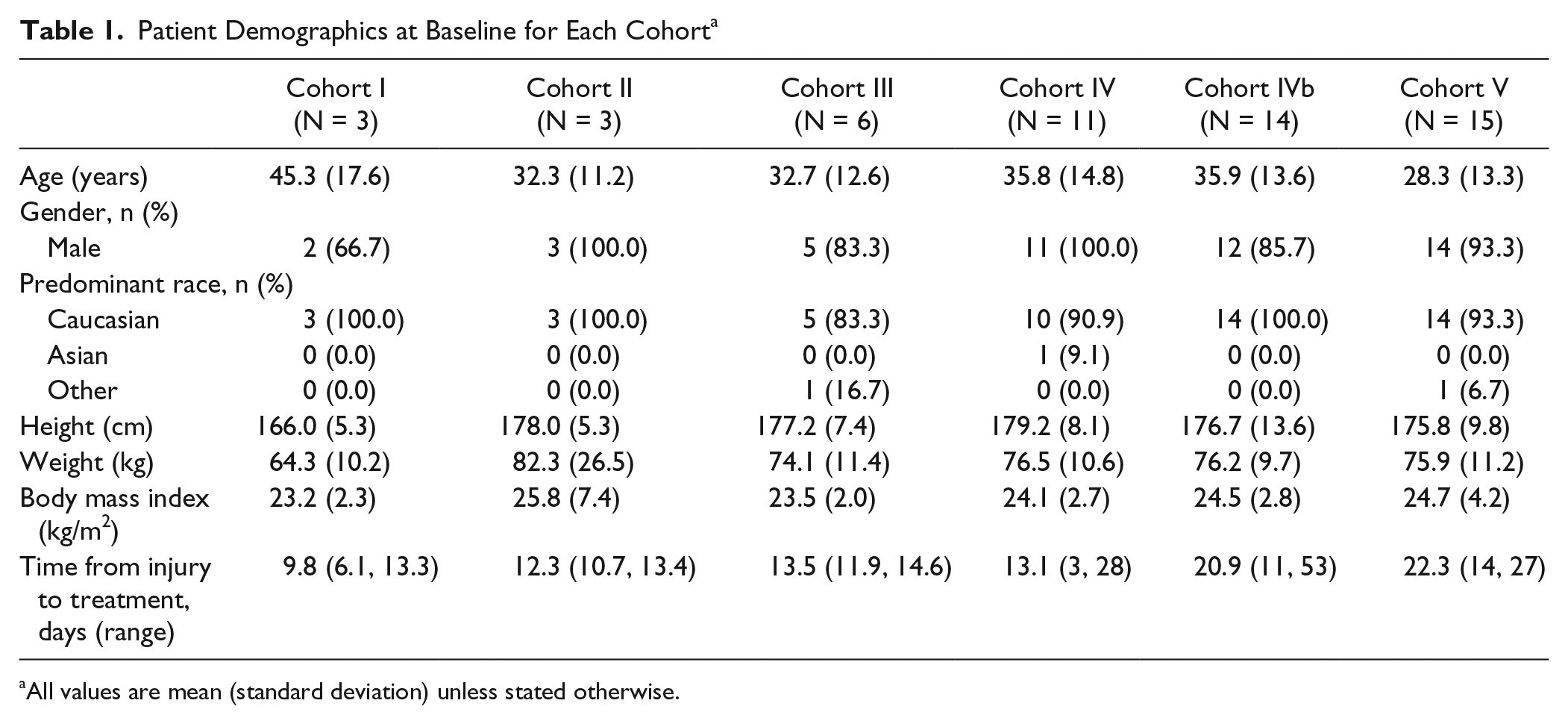
All values are mean (standard deviation) unless stated otherwise.
Seven patients discontinued treatment early but were retained in the study and completed all assessments; reasons for stopping treatment early comprised 5 SAEs and 2 clinically significant AEs (detailed in Table 2B).
Most Frequent AEs by Preferred Term (>33.3% of Patients in Any Cohort) During the Study (A) and Clinically Significant AEs, SAEs, Discontinuations, and Death (B) a .
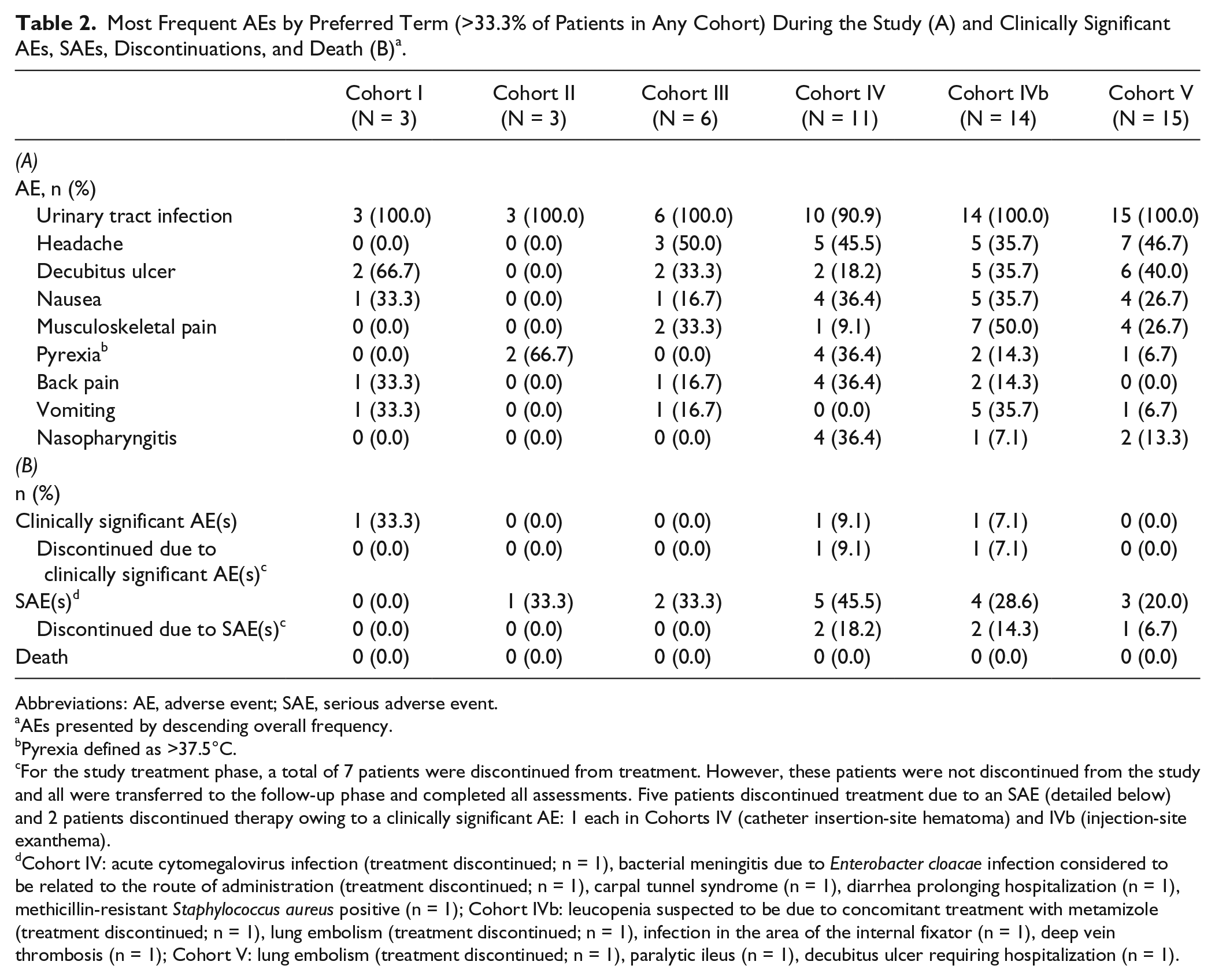
Abbreviations: AE, adverse event; SAE, serious adverse event.
AEs presented by descending overall frequency.
Pyrexia defined as >37.5°C.
For the study treatment phase, a total of 7 patients were discontinued from treatment. However, these patients were not discontinued from the study and all were transferred to the follow-up phase and completed all assessments. Five patients discontinued treatment due to an SAE (detailed below) and 2 patients discontinued therapy owing to a clinically significant AE: 1 each in Cohorts IV (catheter insertion-site hematoma) and IVb (injection-site exanthema).
Cohort IV: acute cytomegalovirus infection (treatment discontinued; n = 1), bacterial meningitis due to Enterobacter cloacae infection considered to be related to the route of administration (treatment discontinued; n = 1), carpal tunnel syndrome (n = 1), diarrhea prolonging hospitalization (n = 1), methicillin-resistant Staphylococcus aureus positive (n = 1); Cohort IVb: leucopenia suspected to be due to concomitant treatment with metamizole (treatment discontinued; n = 1), lung embolism (treatment discontinued; n = 1), infection in the area of the internal fixator (n = 1), deep vein thrombosis (n = 1); Cohort V: lung embolism (treatment discontinued; n = 1), paralytic ileus (n = 1), decubitus ulcer requiring hospitalization (n = 1).
Safety Assessments
Adverse Events
All patients experienced at least 1 AE during the study. There were 581 AEs reported; most were mild in severity, transient, and the majority unrelated to ATI355. The most common AEs were urinary tract infection, headache, and decubitus ulcer (Table 2A). Patients in Cohorts III/IV (2-4 weeks of ATI355 infusion) experienced a mean of 10.8 AEs per patient; patients in Cohorts IVb/V (6 bolus injections of ATI355 over 4 weeks) experienced a mean of 11.7 AEs per patient.
Eight AEs in Cohort III and 7 in Cohort IV were thought to be possibly related to the i.t. continuous infusion method. In comparison, 2 AEs in each of Cohorts IVb/V were considered potentially related to bolus i.t. administration. Headache was the most commonly reported AE considered to be related to i.t. administration and was experienced by 4/23 (17.4%) and 2/29 (6.9%) patients receiving continuous infusion and bolus injections, respectively. Other noteworthy AEs related to the administration route included hematoma at the site of catheter infusion (2 cases for continuous infusion) and medical device complications (1 case in Cohort I of i.t. catheter tear during removal after continuous infusion and 1 case in Cohort IV of infusion line leakage in the area of filter insertion).
Five AEs in Cohorts II/III (none in Cohort I) and 5 AEs in Cohorts IV/IVb (none in Cohort V) were considered by the study investigator to be potentially related to ATI355. In Cohorts II/III, these were headache (n = 2), neck pain (n = 1), leukocytosis (n = 1), and CSF white blood cell count increase (n = 1). In Cohorts IV/IVb, corresponding events were injection-site rash, infection, polyneuropathy (Day 84; predominantly of the axonal type), pruritus, and medical device complication (infusion line leakage in Cohort IV; all n = 1).
Across cohorts, 15 patients reported 16 SAEs during the study (Table 2B), none related to ATI355. One SAE (bacterial meningitis, Cohort IV) was considered related to the administration route; all others were attributed to SCI or concomitant medication. No deaths were reported (Table 2B).
Laboratory Evaluations, Vital Signs, and Other Observations
Deviations from normal values of hematology and blood chemistry parameters were observed in patients across all cohorts, but the AEs associated with these were not clinically significant and did not affect the conduct of the study. No clinically relevant changes in vital signs, ECG, and urine analysis were observed.
Serum PK Assessments
After administration, ATI355 was detectable in serum samples of all patients in Cohorts II to V (Figure 2A, C, and E; Cohorts IV-V only), with increases in exposure proportional to dose increases.
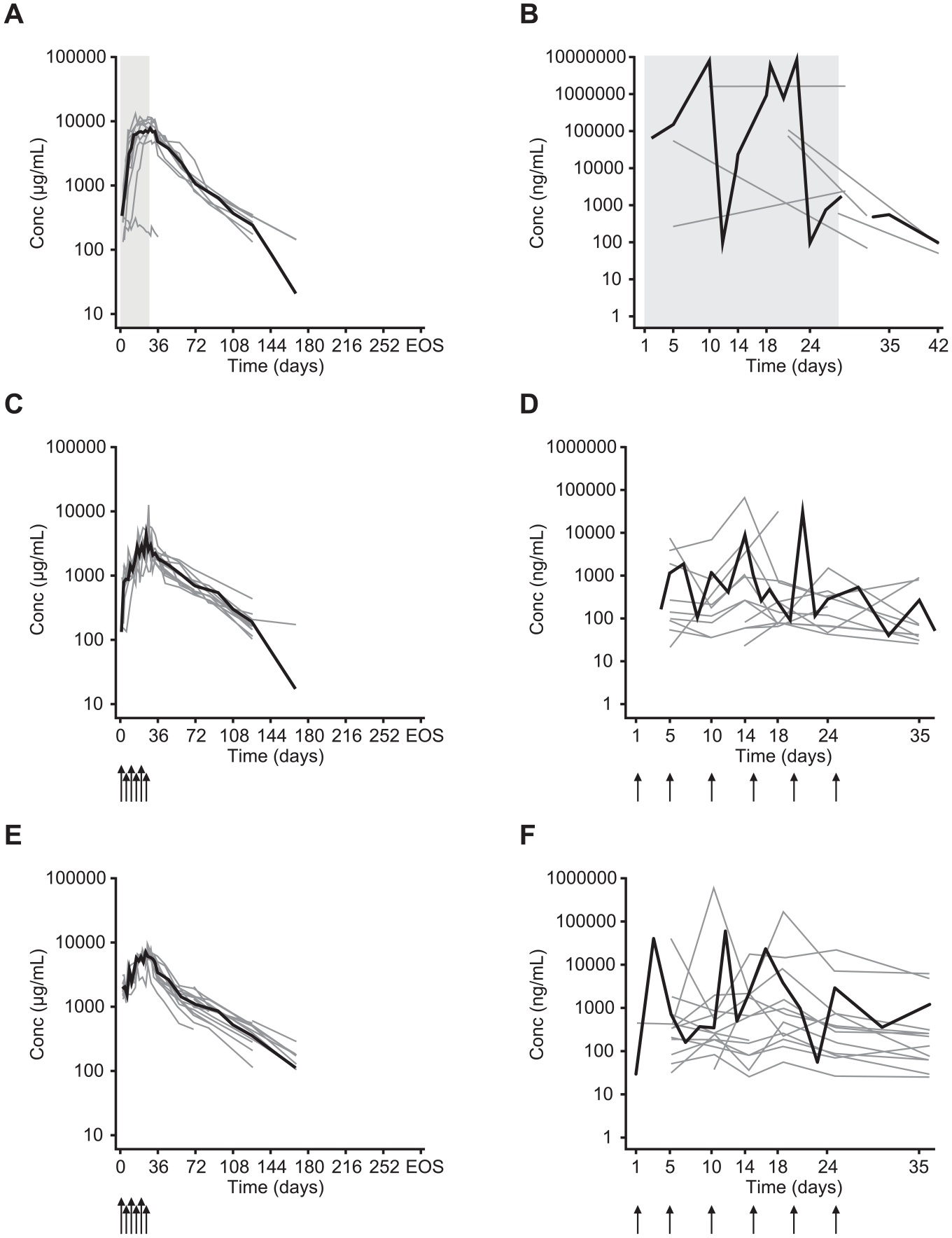
Serum and CSF concentration-time curves of ATI355.
Only 1 patient in Cohort I had detectable serum ATI355 at more than one time point. In Cohort II, the mean serum ATI355 concentration peaked 1 to 3 days after completion of the i.t. infusion, whereas in Cohort III, the concentration peaked during infusion and up to 5 days postinfusion. The mean half-life of ATI355 in serum was approximately 20 to 30 days in Cohorts II/III. In Cohorts IVb/V, serum ATI355 concentrations during the treatment phase were more variable than in the other cohorts (Figure 2C and E).
CSF PK Assessments
After dosing, ATI355 was detected in the CSF samples of all patients, except for 1 patient in Cohort I who also lacked detectable concentrations in serum. Overall, a high degree of variability in the CSF ATI355 concentration was observed after both i.t. infusion and bolus injection (Cohorts IV-V shown as examples in Figure 2B, D, and F). For 8/11 patients in Cohorts I to III who showed a decline in CSF ATI355 concentration, a half-life of approximately 1 to 3 days was assessed; in the remainder, ATI355 remained constant for prolonged periods (ie, up to 42 days postinfusion). These PK observations from Cohorts I to III supported a change from continuous i.t. infusion to i.t. bolus injections every 4 to 5 days in Cohorts IVb/V, with the aim of reducing infection risk and patient discomfort. In Cohorts IVb/V, CSF ATI355 concentrations varied greatly; most patients had concentrations between 100 and 1000 ng/mL (Figure 2D and F). Because of the small sample size per patient, it was not possible to calculate the half-life of ATI355 when administered via i.t. bolus injection. Nevertheless, the observed CSF ATI355 concentrations confirmed the suitability of this regimen.
Serum/CSF Ratio
At Day 35 for Cohorts IVb/V, ATI355 concentration was substantially higher in serum than CSF, with a serum/CSF ratio in the range of 25 to 50 (minimum 0.8, maximum 132.3).
Immunogenicity
No antibodies against ATI355 were detected in any patients, confirming the absence of an immunogenic response.
Neurological Recovery
ISNCSCI Motor Score
In Cohorts I to III, which exclusively investigated paraplegic patients, no changes in mean total motor score (ie, lower extremity motor score [LEMS]) were observed between baseline and study end (data not shown), except in 1 patient who gained 8 points (Cohort II). In the larger cohorts, mean total motor scores for paraplegic patients in Cohorts IV/IVb did not change (data not shown), whereas tetraplegic patients showed a mean increase during the observation period (Cohorts IV, 11 points; IVb, 6 points; V, 6 points). Individual upper motor extremity score (UEMS) and LEMS for Cohorts IV to V are shown in Figure 3. For tetraplegic patients in Cohorts IV/V, any substantial increases in total motor score were gained exclusively through improvement in the UEMS. In Cohort IVb, one tetraplegic patient had a substantial gain in LEMS (from a score of 0 at baseline to a maximum value of 31 at Day 168; final score 16 at end of study).
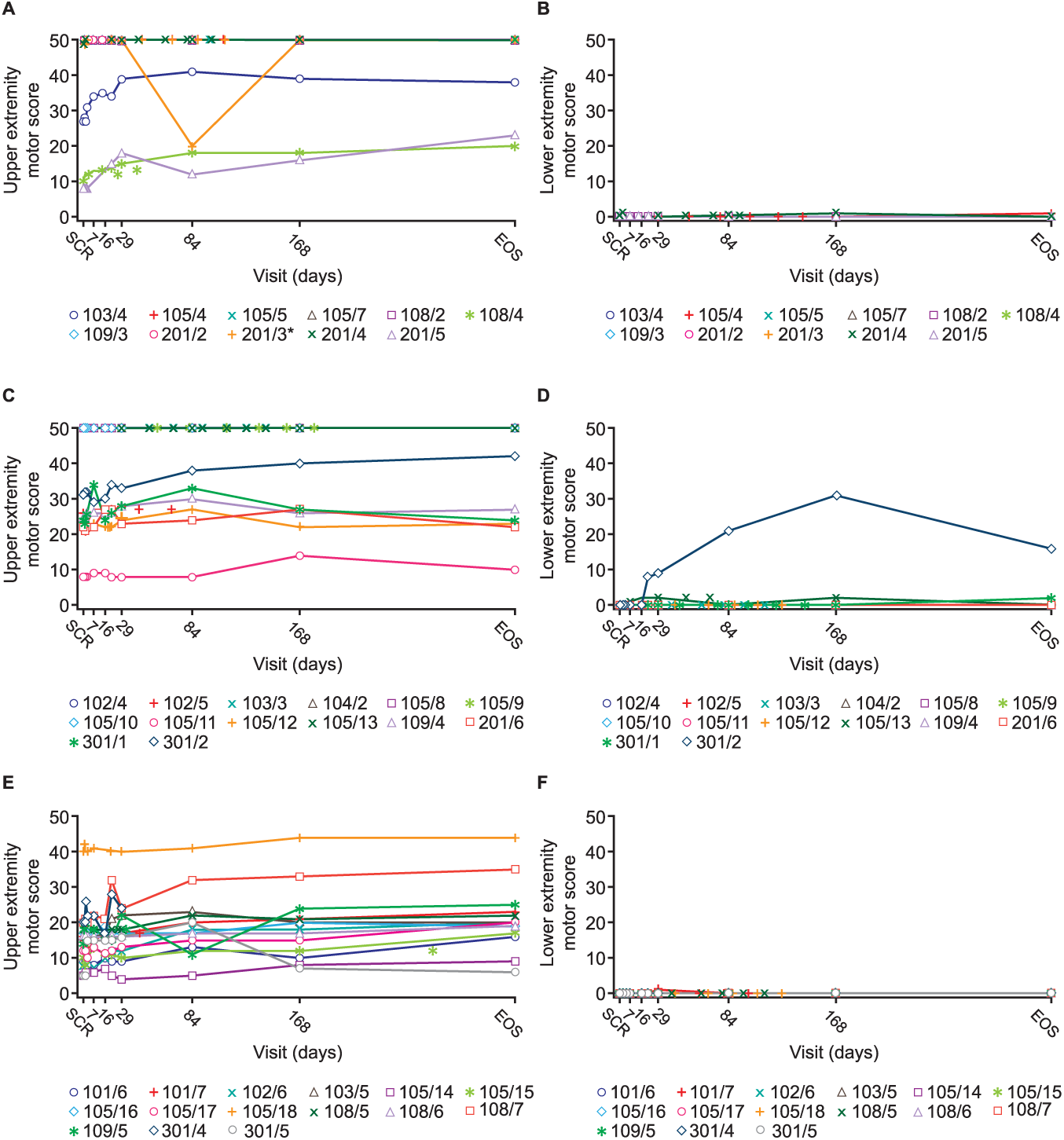
Individual ISNCSCI motor scores over time.
Comparison of the individual total motor score data from baseline to study end (Day 336 [Week 48] in Cohorts IV, IVb, and V) showed that 7/19 evaluable tetraplegic patients had gained >6 points (including improvement in up to 2 myotomes caudal to the neurological level of lesion [NLI]), 3/19 patients >10 points (including improvement in up to 3 myotomes caudal to the NLI), and 1 patient 27 points; the remainder did not show any relevant changes in mean motor scores.
ISNCSCI Sensory Scores
Sensory scores (light touch and pin prick) showed an upwards trend from baseline to study end for all cohorts (Supplementary Figure 1). The greatest improvement was observed in tetraplegic patients in Cohort IVb, with a 16.4-point increase for the pin prick and 21.7 for the light touch mean scores from baseline to study end.
AIS
No patients in Cohort I showed an improvement in AIS grade. Overall, 1 patient in each of Cohorts II/III improved to a higher AIS grade (Table 3). The conversion at Day 3 from AIS A to AIS B in Cohort II was unlikely an effect of ATI355 (ie, not associated with the expected mechanism of action of ATI355); this patient later converted to AIS C by Day 84, gaining 8 points in motor score. Of the tetraplegic patients, 3 in Cohort IVb and 4 in Cohort V showed a conversion from complete SCI (AIS A) to incomplete SCI. Of note, only 1 conversion (in Cohort IVb) occurred within the first 30 days of the study, with a further 3 conversions occurring by Day 84, 2 between Days 84 and 168, and 1 before the end of the study (Day 336). By the end of the study, of the 19 evaluable tetraplegic patients, 5 (2 in Cohort IVb and 3 in Cohort V) had converted from AIS A to AIS B and 2 (1 in both Cohorts IVb/V) from AIS A to AIS C (Table 3).
Timing of Conversion in ASIA Impairment Scale (AIS) Grades per Cohort (Shown as Number of Patients Classified at Each Grade by Time Point).
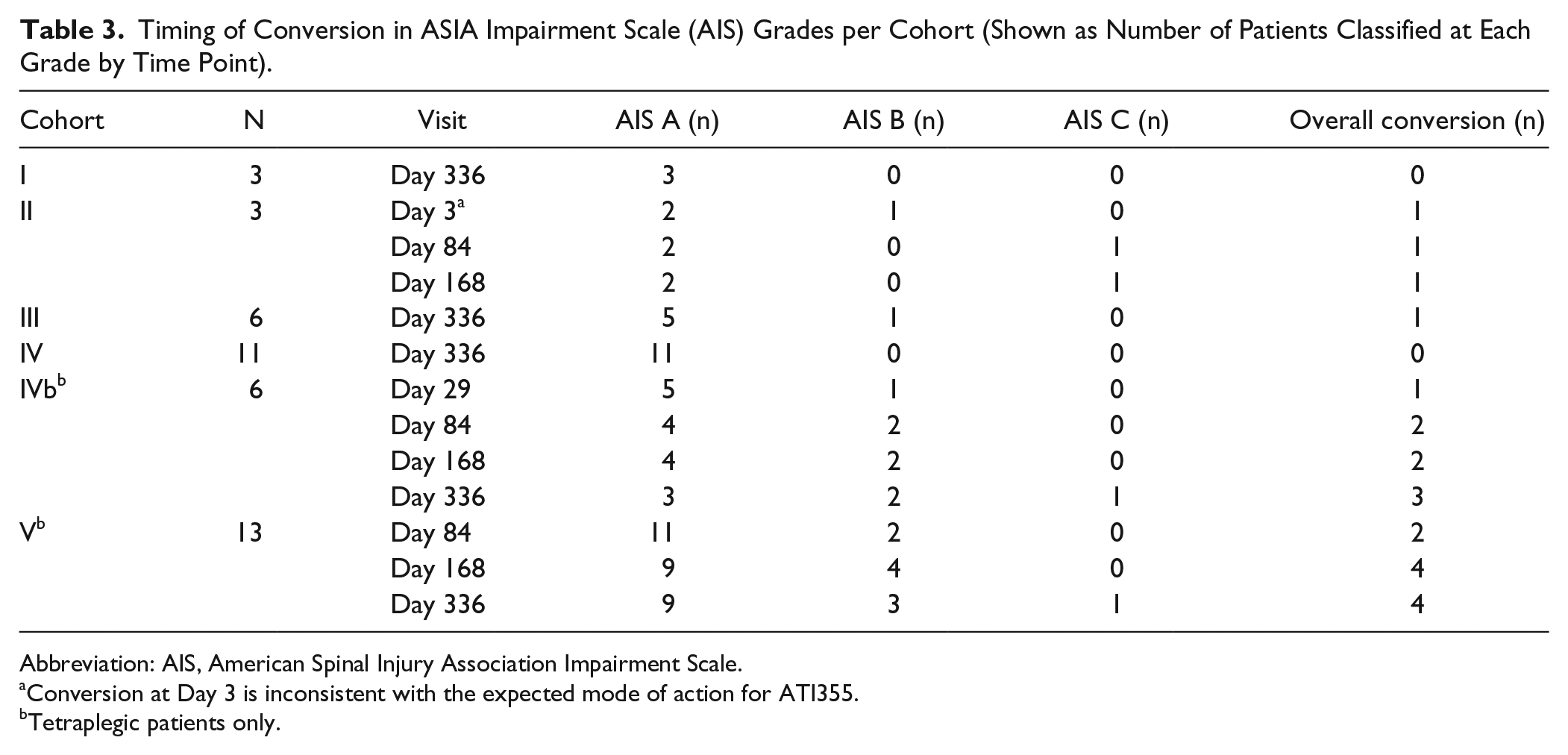
Abbreviation: AIS, American Spinal Injury Association Impairment Scale.
Conversion at Day 3 is inconsistent with the expected mode of action for ATI355.
Tetraplegic patients only.
Electrophysiological Tests
No relevant changes in tibial NCV were observed in any cohort. Owing to the nature of the SCI, it was not possible to derive SSEP and MEP responses for the majority of patients. Of the patients with measurable responses, there were no significant changes in SSEP (stimulating the tibial [n = 2] and the ulnar nerve [n = 11 with cervical lesions]; Cohorts IV-V]) or MEP amplitudes/latencies (n = 2; tibialis anterior muscle).
Discussion
This first-in-man study assessed the feasibility, acute safety and tolerability, PK, and neurological recovery of i.t.-administered ATI355, an anti-Nogo-A recombinant human antibody, in the treatment of acute SCI. Nogo-A is one of several factors that inhibits spontaneous neural regeneration in vitro, and animal studies have shown that its blockade allows enhanced sprouting and reconnection of spared fibers, and improved functional recovery.15,17,20 Thus far, clinicians have been hesitant to use i.t. drug administration in acute SCI because direct drug administration into the CSF is more challenging than common administration routes. However, i.t. dosing bypasses the blood-brain barrier, which is required when targeting antibodies in a sufficiently high dosage to the CNS, and may improve tolerability by permitting delivery of smaller doses than would be required intravenously. 29
This study demonstrated that ATI355 was generally well tolerated in patients with acute traumatic paraplegia and tetraplegia following continuous i.t. infusion or repeated i.t. bolus injections. Reported AEs were broadly consistent with conditions commonly observed in patients with SCI,30-33 were mostly mild in severity, and the majority considered unrelated to ATI355. One case of polyneuropathy was reported on Day 84 to be potentially related to ATI355, and while it is not possible to be conclusive regarding causality in this relatively small sample size with no placebo group, polyneuropathy has been documented previously as a consequence of cervical SCI. 34
The most reported AE considered related to i.t. administration was headache, a common complication following a puncture of the spinal dura mater due to potential CSF leakage with consequential lowering of CSF pressure. 35 Two additional cases of headache were attributed by study investigators to ATI355, but in retrospect were likely to have been related to the route of administration. Quantifying differences in safety between continuous i.t. infusion and bolus injection was restricted by few patients receiving ATI355 via each route of administration. Regardless, the risk of infection when administering to an immune-privileged site such as the CSF should not be overlooked; we believe intermittent dosing may be advantageous in this regard. Bacterial meningitis occurred in a patient receiving ATI355 by continuous i.t. infusion and was likely related to the administration route. Technical complications related to use of an indwelling catheter for continuous i.t. infusion, including catheter breakage during withdrawal and infusion line leakage in the filter insertion area, were also observed. Practical concerns with continuous i.t. infusion, such as immobilizing the patient for prolonged periods, interferes with rehabilitative interventions. Consequently, continuous i.t. administration was substituted with intermittent i.t. bolus injection. The incidence of headache in Cohorts IVb/V, which received i.t. bolus injections, was slightly higher compared with the continuous-application cohorts; no other AEs or SAEs related to bolus injection were identified.
Many therapeutic antibodies induce the production of human anti-human antibodies, a process influenced by the duration of treatment and whether the antibody is humanized or fully human. 22 Consistent with the lower risk associated with a fully human antibody, no anti-ATI355 antibodies were detected in blood samples, confirming a low risk of immunogenicity. Other contributory factors may include the high quality of the antibody, the short treatment duration, or the i.t. administration route.
The PK data demonstrate that both continuous i.t. infusion of 15 mg/day ATI355 over 14 to 28 days (Cohorts III/IV) and 6 i.t. bolus injections (22.5 or 45 mg/injection) over approximately 24 days (Cohorts IVb/V) were suitable to maintain relevant ATI355 CSF concentrations for a minimum of 4 to 5 weeks. Serum concentrations were proportional to dose; therefore, patients in Cohort IV infused for 28 days had the greatest exposure to ATI355. Patients who received 6 i.t. bolus injections received comparatively lower doses; however, the dose and injection volume in these cohorts was not increased for safety reasons.
As expected for an IgG4 monoclonal antibody (serum half-life: ~3 weeks), 36 and as observed following intraventricular administration of the IgG monoclonal antibodies trastuzumab 37 and rituximab, 38 relevant ATI355 concentrations persisted longer in serum than in CSF.
Intrasubject variability in ATI355 CSF concentrations was observed after both continuous i.t. infusion and bolus injection. Potential causes may include patient body position during sample collection by lumbar puncture, possible stenosis/cord swelling at the lesion site preventing even distribution across the CSF compartment, or altered CSF absorption due to trauma-induced hemorrhage.
CSF concentrations were mostly around 0.1 µg/mL (Figure 2B, D, and F). Preclinical data from in vitro binding assays and activity studies suggest that this would be sufficient to effectively block Nogo-A and allow neurite outgrowth (see Supplementary Preclinical Appendix). An important consideration is whether ATI355 injected at the lumbar site distributes at sufficient concentrations to the spinal cord lesion and to CNS regions rostral to the lesion. Preclinical experiments in primates suggest good distribution of ATI355 after bolus (lumbar injection; unpublished data) or i.t. infusion (cervical spinal cord) 25 into more distant sites such as the thoracic spinal cord and brain. Additionally, Kuttler et al 39 have described the distribution of large biomolecules in the CSF after lumbar i.t. injection using biophysical fluid-dynamic models. However, the potential impact of persistent spinal canal encroachment and cord edema on CSF dynamics is complex and was not assessed here.
In this study, no patients experienced a clinical deterioration in motor and sensory scores, further supporting the safety of ATI355. One paraplegic patient—who would have had normal UEMS—gained a substantial increase in motor score (+8 points in LEMS). The absence of demonstrable neurological improvement in paraplegic patients is not unexpected owing to ISNCSCI’s limited ability to detect improvements in motor function at a neurological level in the thoracic region. One-year data from the European Multicenter Study about SCI (EMSCI) from patients with thoracic complete SCI demonstrated that spontaneous recovery in LEMS rarely occurs and is more likely in subjects with low thoracic SCI—of which there were few in Cohorts IV/IVb (T10-T12: 2/8 and 2/7 patients, respectively). 40
Of the 19 evaluable tetraplegic patients, 7 gained ≥6 points, 3 ≥10 points, and 1 ≥20 points over 48 weeks of follow-up. For all but 1 of these patients, the gain in total motor score was entirely contributed by an increase in UEMS, as expected after cervical lesion. Retrospective, longitudinal data (from EMSCI/Sygen study) report an average spontaneous recovery in UEMS of 10 to 11 points over 1 year in patients with C5-C7 lesions. 41 Almost half of the improvements reported in the EMSCI/Sygen study were gained in the first 8 weeks following SCI. Eligible patients in the current study were permitted to delay entry by up to 28 or 60 days post-SCI, and thus the current findings may not capture the full extent of recovery in the enrolled tetraplegic patients. Despite the large gains in motor scores in some of the tetraplegic patients, overall mean increases in motor scores were 11 points in Cohort IV and, more modestly, 6 points in Cohorts IVb/V. It is noteworthy, however, that the neurological improvements observed here occurred in a time window (3-12 months after SCI) when spontaneous improvements are very rare and, if they do occur, small. Although mean scores are important assessors of overall benefit, caution should be taken when interpreting them in small patient cohorts, such as in this study, owing to the potential for skewing arising from wide intersubject variability. Without a comparator group, it is difficult to attribute the observed clinical improvement to ATI335. Placebo-controlled trials are required to further evaluate the safety and tolerability, and ascertain the clinical efficacy of, ATI335.
All cohorts showed changes in ISNCSCI sensory scores from baseline to study end that were either comparable to or exceeded those reported in historical controls. 40 In the EMSCI dataset, sensory levels exhibited only minor changes and included both improvement and deterioration during the first year following injury. 40 Overall, the different regimens of i.t. drug application and ATI355 treatment did not negatively affect neurological status.
This study was restricted to patients with complete SCI (ie, reduced extent of spontaneous recovery) owing to potential safety concerns. Additionally, before the study began, it was considered challenging to predict recovery in incomplete human SCI. Prediction models (unbiased recursive partitioning) for patients with acute complete and incomplete SCI were recently developed and validated. Inclusive protocol designs now allow enrollment of different patient cohorts, basing eligibility for inclusion on patients’ individual predicted outcomes.42-44 These protocols allow parallel inclusion of patients with complete and incomplete SCI and improve both the sensitivity of the measured endpoints and the power of a study. Furthermore, the combination of related neurological and functional outcome measures can be applied to reveal the clinical relevance of outcome measures.45-47 Our study results lay the groundwork for Phase II/III studies and the application of inclusive protocols and advanced prediction models.
In summary, this study represents the first-in-man investigation of i.t. antibody delivery for the treatment of acute SCI. We demonstrate that ATI335 is well tolerated, and i.t. delivery, particularly via bolus injection, is a viable mode of drug administration in acute SCI. The small number of subjects and the open-label nature of this study limit our ability to draw conclusions regarding efficacy. Larger-scale, randomized, placebo-controlled trials are required to further investigate the efficacy of ATI355 in improving neurological recovery after SCI.
Supplementary Material
Supplementary Material, NOV0119_Supplementary_tables – First-in-Man Intrathecal Application of Neurite Growth-Promoting Anti-Nogo-A Antibodies in Acute Spinal Cord Injury
Supplementary Material, NOV0119_Supplementary_tables for First-in-Man Intrathecal Application of Neurite Growth-Promoting Anti-Nogo-A Antibodies in Acute Spinal Cord Injury by Klaus Kucher, Donald Johns, Doris Maier, Rainer Abel, Andreas Badke, Hagen Baron, Roland Thietje, Steven Casha, Renate Meindl, Baltazar Gomez-Mancilla, Christian Pfister, Rüdiger Rupp, Norbert Weidner, Anis Mir, Martin E. Schwab, and Armin Curt in Neurorehabilitation and Neural Repair
Supplementary Material
Supplementary Material, Supplementary_Methods_29Aug2017 – First-in-Man Intrathecal Application of Neurite Growth-Promoting Anti-Nogo-A Antibodies in Acute Spinal Cord Injury
Supplementary Material, Supplementary_Methods_29Aug2017 for First-in-Man Intrathecal Application of Neurite Growth-Promoting Anti-Nogo-A Antibodies in Acute Spinal Cord Injury by Klaus Kucher, Donald Johns, Doris Maier, Rainer Abel, Andreas Badke, Hagen Baron, Roland Thietje, Steven Casha, Renate Meindl, Baltazar Gomez-Mancilla, Christian Pfister, Rüdiger Rupp, Norbert Weidner, Anis Mir, Martin E. Schwab, and Armin Curt in Neurorehabilitation and Neural Repair
Supplementary Material
Supplementary Material, Supplementary_preclinical_appendix_to_submit – First-in-Man Intrathecal Application of Neurite Growth-Promoting Anti-Nogo-A Antibodies in Acute Spinal Cord Injury
Supplementary Material, Supplementary_preclinical_appendix_to_submit for First-in-Man Intrathecal Application of Neurite Growth-Promoting Anti-Nogo-A Antibodies in Acute Spinal Cord Injury by Klaus Kucher, Donald Johns, Doris Maier, Rainer Abel, Andreas Badke, Hagen Baron, Roland Thietje, Steven Casha, Renate Meindl, Baltazar Gomez-Mancilla, Christian Pfister, Rüdiger Rupp, Norbert Weidner, Anis Mir, Martin E. Schwab, and Armin Curt in Neurorehabilitation and Neural Repair
Footnotes
Acknowledgements
We thank Edwige Chokote and Denise Sickert for immunogenicity testing, Fabienne Deckert-Salva for assistance with PK sample management, and Orpheus Mach who acted as study coordinator at the Murnau site, which enrolled one third of the study population. We also thank A. Vitaliti and M. A. Brauchle for the ATI355 characterization data that are included in the Supplementary Appendix.
Supplementary material for this article is available on the Neurorehabilitation & Neural Repair website along with the online version of this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Klaus Kucher, Baltazar Gomez-Mancilla, Christian Pfister, and Anis Mir are employees and stockholders of Novartis Pharma AG, Basel, Switzerland. Donald Johns was an employee and stock holder of Novartis Pharmaceuticals Corporation at the time of this work. He is no longer employed at Novartis (current affiliation: AxonGuidance, MA, USA) and is no longer a stock holder of Novartis. Martin E. Schwab is an employee of the University of Zurich and the Swiss Federal Institute of Technology (ETH) Zurich. He is a cofounder and president of NovaGo Therapeutics Inc, a company dedicated to the development of novel therapies for neurological diseases. Rüdiger Rupp, Norbert Weidner, and Armin Curt have acted as consultants to Novartis. Doris Maier, Rainer Abel, Andreas Badke, Hagen Baron, Steven Casha, Renate Meindl, and Roland Thietje have no competing interests to report.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Novartis Institutes for BioMedical Research, Basel, Switzerland. Medical writing and editorial assistance was provided by Julianna Solomons, PhD, at Fishawack Communications Ltd, Abingdon, Oxon, UK; this service was funded by Novartis Pharma AG, Basel, Switzerland.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.