
Abstract
Objective. Genetic variations in the dopamine (DA) system are associated with cortical-striatal behavior in multiple populations. This study assessed associations of functional polymorphisms in the ankyrin repeat and kinase domain (ANKK1; Taq1a) and catechol-O-methyltransferase (COMT; Val158Met) genes with behavioral dysfunction following traumatic brain injury (TBI). Participants. This was a prospective study of 90 survivors of severe TBI recruited from a level 1 trauma center. Main measures. The Frontal Systems Behavior Scale, a self- or family report questionnaire evaluating behavior associated with frontal lobe dysfunction, was completed 6 and 12 months postinjury. Depression was measured concurrently with the Patient Health Questionnaire-9. Study participants were genotyped for Val158Met and Taq1a polymorphisms. Results. No statistically significant behavioral differences were observed by Taq1a or Val158Met genotype alone. At 12 months, among those with depression, Met homozygotes (Val158Met) self-reported worse behavior than Val carriers (P = .015), and A2 homozygotes (Taq1a) self-reported worse behavior than A1 carriers (P = .028) in bivariable analysis. Multivariable models suggest an interaction between depression and genetic variation with behavior at 12 months post-TBI, and descriptive analysis suggests that carriage of both risk alleles may contribute to worse behavioral performance than carriage of either risk allele alone. Conclusion. In the context of depression, Val158Met and Taq1a polymorphisms are individually associated with behavioral dysfunction 12 months following severe TBI, with preliminary evidence suggesting cumulative, or perhaps epistatic, effects of COMT and ANKK1 on behavioral dysfunction.
Introduction
Understanding and managing traumatic brain injury (TBI) can be challenging because people with similar injury profiles can experience different cognitive, emotional, and behavioral outcomes.1-4 Rehabilomics is a conceptual framework from which to investigate these diverse outcomes by examining the complex interplay between personal, biological, and psychosocial factors present in the context of TBI.5,6 Rehabilomics is unique in its inclusion of personal biological factors, such as genetic variation and serum biomarkers, which may contribute directly to TBI outcomes or interact with other biological and functional factors to affect outcomes. As such, the rehabilomics framework can inform study designs for understanding biological mechanisms underlying various outcomes and neural recovery after TBI. The framework also provides a theoretical basis for developing personalized-medicine approaches to neurorehabilitation after TBI. As an example, our previous work has used a rehabilomics approach to study heterogeneity in cognitive deficits following TBI, incorporating personal (genetic) and biological (sex) factors. 7 Furthermore, genetic variation influences on personal traits and psychiatric disorders is increasingly recognized as relevant to clinical practice. 8 Similar approaches can be used to study other complex TBI outcomes, including behavioral problems, with the goal of learning how best to design biologically tailored rehabilitation strategies.
It is known that 54% of individuals with moderate/severe TBI report behavioral problems that persist for years postinjury,9,10 including aggression, disinhibition, amotivation, and difficulty planning and executing actions.11,12 Behavioral problems result from the complex interactions among cognition (eg, cognitive control), emotional state (eg, depressive symptoms), and personal factors (eg, genetics), manifesting primarily in response to environmental stimuli. 13 We have shown previously that the dopamine (DA) system is highly susceptible to dysfunction following TBI. 14 Clinically, DA agonists can improve outcome15,14 and have neurorestorative effects in in vivo experimental TBI models,16,17 suggesting that TBI results in a functional hypodopaminergia, although mechanisms by which this occurs are still largely unknown. DA modulated processing can affect cognition via projections to the mesocortical system (prefrontal cortex [PFC] and medial frontal cortices), emotion via amygdala and cingulate projections, and depression and amotivation by projections to the hippocampus and ventral striatum (VS).18-20 The DA system is under complex regulatory control by afferent regions and by the interplay between tonic and phasic elements of neurotransmission, where tonic DA levels modulate stimulus-driven phasic DA release. 21 The literature underscores the importance of DA systems, in humans and animal models, to PFC-centered behaviors such as aggression,22,23 impulsivity,24,25 and executive function.26,27 DA also influences other PFC-centered constructs such as cognition, which when impaired, contributes to behavioral problems. Clinical dopaminergic therapy studies involving attention-deficit/hyperactivity disorder 28 and Parkinson’s disease 29 demonstrate that DA system modulation can improve problematic behaviors.
The PFC uses cortical striatal afferents to govern elements of executive function that include aspects of social cognition and emotional regulation that influence behavior. 30 Furthermore, the PFC has the capacity for top-down regulatory control over ascending DA modulatory systems in a manner specific to the environmental stimuli and stressors at hand31,32; so examining variation within candidate genes involved in these connected DA regions could inform how these regions regulate behavior.33,34 Whereas numerous genes regulate DA system control, we have chosen 2 genes with well-described functional polymorphisms that have been highlighted as primary genes of importance in TBI-specific outcomes research. 35 These 2 candidate genes are known as COMT (catechol-O-methyltransferase) and ANKK1. COMT is the enzyme primarily responsible for DA metabolism in the PFC and is linked to impulsivity and aggression among individuals with schizophrenia. 36 Within COMT, there exists a well-studied functional polymorphism called Val158Met (rs4680). The Val allele has 4 times greater enzyme function than the Met allele, which leads to lower DA levels at cortical synapses but increased phasic responses at subcortical synapses. 37 Compared with standard mouse models, COMT overexpressing mice exhibit differences in DA release in the VS, implicating a role for striatal DA metabolism as well. 38 The Taq1A polymorphism (rs1800497) in the ANKK1 gene, associated with DA type-2 receptor D2, 39 is implicated with impulsivity in healthy populations 40 and childhood aggression, 41 both of which are associated with DA system disruption. Taq1A polymorphisms also are associated with D2 presynaptic/postsynaptic receptor densities 42 and are highly expressed in subcortical regions. 43 Studies report that Taq1A A1 carriers have lower receptor densities than A2 homozygotes,42,44,45 but A2 carriers may have lower receptor densities in the context of depression. 46 Thus, the A2 allele may actually impart a greater risk for poor DA modulation that differs from those for healthy populations. Whereas other genetic components of the DA system may influence outcomes, COMT and ANKK1 are currently the only genes with both a strong mechanistic rationale and previously documented associations with other TBI outcomes (cognition) in the literature.47-49
Similarly, some studies indicate that genetic risk relationships between both Val158Met50,51 and Taq1A 52 and behavior are only present in the context of a moderating stressor. Disrupted subcortical and PFC activity can occur in the presence of a chronic stressor such as posttraumatic stress disorder (PTSD) 53 or depression 54 to decrease cognitive control over behavior, leading to increased reliance on emotion-based decision making and the emergence of poor behaviors similar to those observed with TBI. This phenomenon has been characterized as a switch from top-down cognitive control, in which the PFC controls subcortical regions to plan and execute a decision, to a state of bottom-up emotional control, where subcortical regions involving emotion and reward systems function with reduced PFC regulation. 55 This framework suggests that cognitive control interacts with emotional state to contribute to behavioral problems. Chronic rodent stress models lead to anxiety and despair-like behaviors that are associated with decreased DA neuron activity. 56 Post-traumatic Depression (PTD) rates are ~50% during the first year post-TBI. 57 PTD is associated with poor behavior postinjury 9 and may trigger a positive feedback loop of behavioral problems and depressive symptoms across this time period. 58
Whereas previous work has focused on how DA genetics can influence cognition after TBI7,47 only one other study exists in the TBI literature examining DA system genetics and behavioral dysfunction—specifically aggression. 59 Because both Val158Met and Taq1A polymorphisms are associated with PFC and VS DA neurotransmission and related behavior, further TBI investigation is warranted. Thus, we examined how DA genetic variation influences behavior after TBI, both independently and in the context of a chronic stressor, specifically PTD. Based on previous literature, we hypothesized that Val158Met and Taq1A polymorphisms would be associated with PFC-centered behavior. Furthermore, we hypothesized that genetic predilection to relatively increased PFC DA levels, associated with the COMT gene Met allele, would be associated with poorer behavior post-TBI. Given ANKK1 gene associations with stress-inducing conditions such as PTSD,52,60 we hypothesized that ANKK1 genetic variation would influence behavior after TBI, particularly among those with PTD, wherein reduced mesostriatal DA neuron phasic firing and PFC overactivity 56 may drive an imbalance with corticostriatal tonic-phasic DA modulation. 61 Whereas A1 carriers can have worse behavioral and cognitive outcomes in healthy and mild TBI (mTBI) populations, our data characterizing those with severe TBI suggest that A2 homozygotes have worse cognitive outcomes. 47 This study aims to clarify which alleles impart risk for poor behavioral outcomes in a moderate-to-severe TBI population. Because PTD often emerges within the first 6 months postinjury, 57 we expected the potential chronic stress effects of PTD on the DA gene–behavior relationships to be most evident at 12 months postinjury.
Methods
Participants were recruited from inpatient and outpatient centers at the University of Pittsburgh Medical Center as part of a larger TBI study approved by our institutional review board. Enrollment criteria included a nonpenetrating severe TBI (admission Glasgow Coma Scale [GCS] ≤8), a computed tomographic scan with evidence of intracranial injury, and age 16 to 75 years. Participants with documented evidence of hypoxia (>30 minutes) occurring prior to admission were excluded. Behavioral data at either 6 or 12 months postinjury were available for 97 participants. Due to concerns regarding racial stratification 62 and differences in allele frequency distribution between races in Taq1A 63 and Val158Met, 64 this study was then restricted to self-reported white individuals, leaving a final cohort of 90 unique participants (6 months, n = 69; 12 months, n = 69; both 6 and 12 months, n = 48). Supplemental Figure 1 shows further participant breakdown. Demographic information was obtained from medical records and/or participant/caregiver interview. The best GCS within 24 hours postinjury was used for analysis, given its discriminative ability when examining outcomes. 65 Assessors were blinded to genotype status.
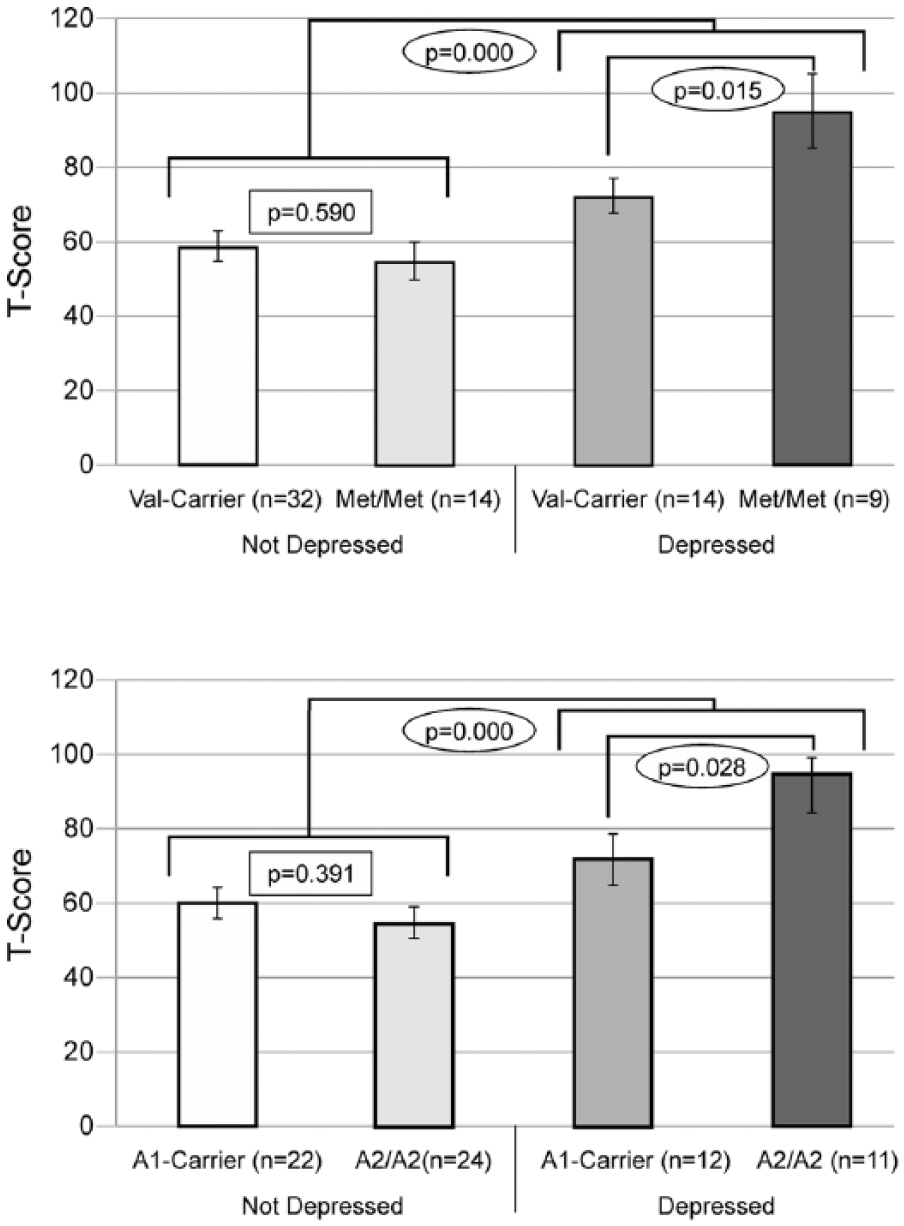
12-Month results: carrier status and depression groups were compared separately for both Val158Met and Taq1a. Those with PTD exhibited significantly worse outcomes regardless of genetic status, and statistically significant differences by carrier status were only noted between those with PTD. Similar nonsignificant trends were also observed at 6 months (data not shown).
Behavior Assessment
The Frontal Systems Behavior Scale (FrSBe) is a validated assessment of behaviors associated with damage to the frontal lobes and includes an overall score and 3 subscale scores: disinhibition, apathy, and executive dysfunction 66 ; both the self-report and family-report versions were administered when possible. 67 Questions were scored with regard to current behavior (after injury) and preinjury. Standardized FrSBe scoring yields norm-based T-scores adjusted for age, sex, and education. Higher T-scores indicate more problem behaviors.
Depression Assessment
The Patient Health Questionnaire-9 is a validated self-report symptom-based questionnaire based on the DSM-IV (Diagnostic and Statistical Manual of Mental Disorders, 4th ed) criteria for depression and is validated for use after TBI. 68 Participants were categorized at 6 and 12 months as having PTD if endorsing ≥5 symptoms, at least one of which was a cardinal symptom of depression (depressed mood or anhedonia). History of premorbid psychiatric disorders, including depression, bipolar disorder, and/or anxiety disorder, was collected from interview or medical chart review.
Genotyping
DNA was isolated from blood using a simple salting out procedure 69 and genotyped for COMT (rs4680, Val158Met) and ANKK1 (rs1800497, Taq1A variant). COMT Val158Met (rs4680) was genotyped using TaqMan allele discrimination technology and available 5′ exonuclease Assay-on-Demand TaqMan assays (Applied Biosystems). For ANKK1 Taq1A (rs1800497) genotyping, amplified DNA underwent 30 cycles of denaturation at 95°C for 1 minute, annealing at 58°C for 30s, and extension at 72°C for 1 minute to amplify the 459bp product, which was then exposed to TaqI restriction endonuclease to perform restriction fragment length polymorphism analysis. Digested products were electrophoresed on a 3% agarose gel, stained with ethidium bromide for DNA band detection, and assigned a genotype based on presence/absence of original or cut DNA fragments. Primers used were 5′-CCGTCGACCCTTCCTGAGTGTCATCA-3′ and 5′-CCGTCGACGGCTGGCCAAGTTG TCTA-3′. Two individuals blinded to phenotype data 69 called each genotype, and discrepancies were resolved by examining the raw data and rerunning samples if necessary. We grouped participants into Met homozygotes versus Val carriers, and A1 carriers versus A2 homozygotes based on allele frequency, function, or previous studies in the literature.
Data Analysis
Statistical analyses were completed using SPSS (version 22). Mean, median, SD, and standard error were calculated when appropriate, and categorical data were reported as frequencies. Behavior and demographic data were compared using Mann-Whitney U, ANOVA, Kruskall-Wallis test, T-tests, or χ2 tests as appropriate. Group differences considering carrier and depression status were assessed using ANOVA and post hoc analysis with Fisher’s least significant difference. We tested both Val158Met and ANKK1 in separate multiple regression models controlling for preinjury psychiatric disorders, antidepressant use, and behavior before injury (FrSBe before total T-score). Partial η2 values were reported to determine the amount of variance in behavior captured with each variable tested in each multivariable model.
Results
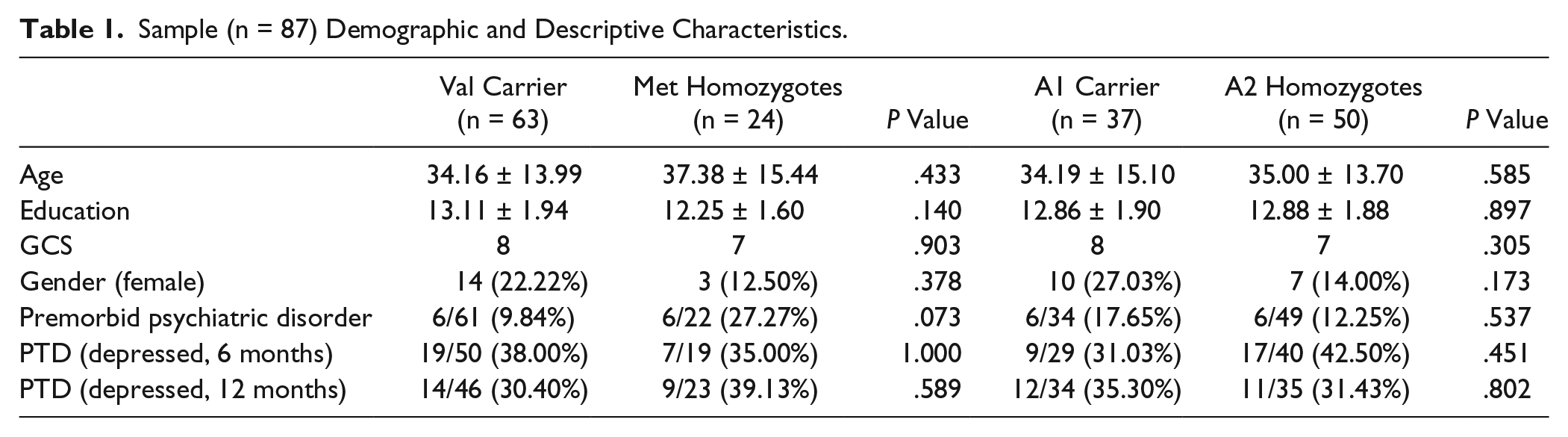
Demographic data for the entire sample are presented in Table 1. No significant differences by genotypes were identified for any descriptive factor. Bivariable analyses yielded no significant differences in self-reported behavior between Met homozygotes and Val carriers or between A1 carriers and A2 homozygotes at 6 months or 12 months postinjury. The sample was in Hardy-Weinberg equilibrium for each variant studied.
Sample (n = 87) Demographic and Descriptive Characteristics.
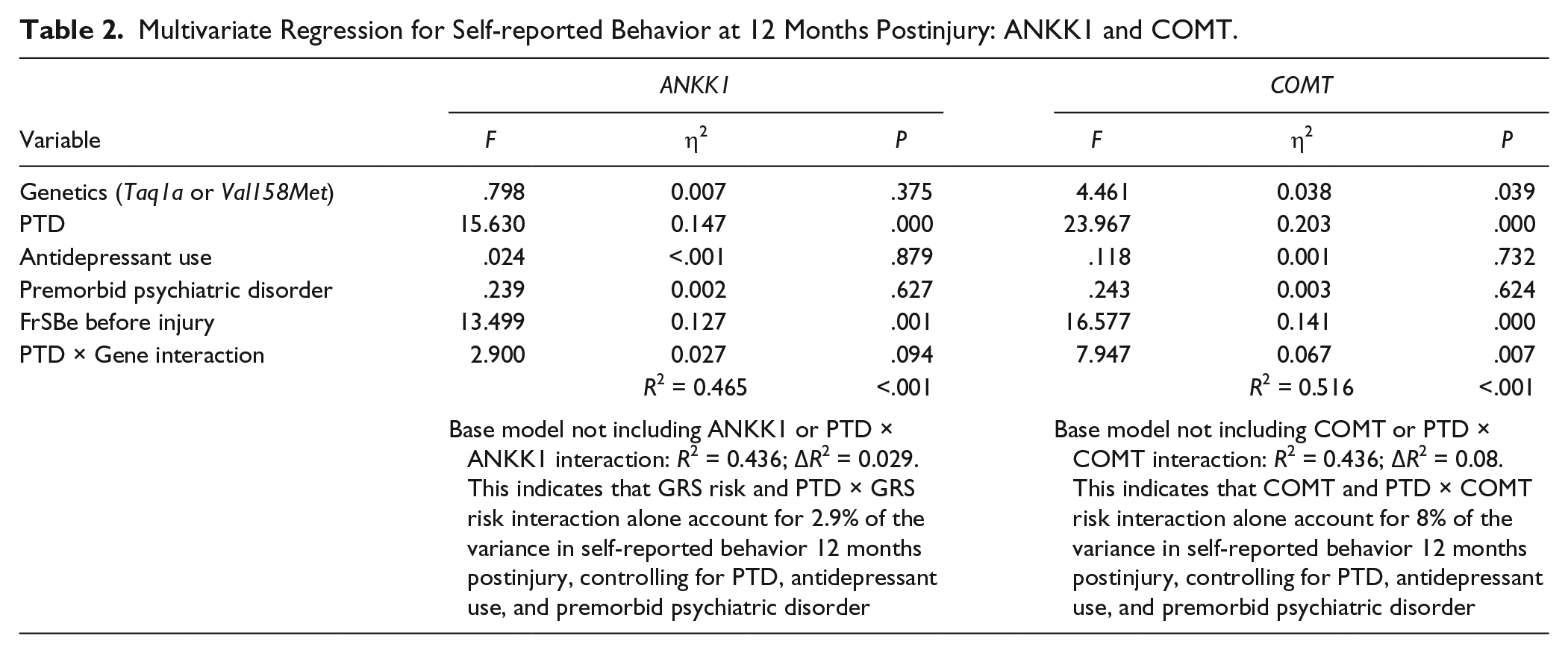
When examining relationships between genes and behavior by PTD status, we found a significant difference in behavior by Val158Met status only among those with PTD at 12 months (P = .028) but not at 6 months (P = .073) postinjury. We found similar results for Taq1A status at 12 months (P = .028) but not 6 months (P = .884) postinjury. Figure 1 illustrates no differences between Val carriers and Met homozygotes among those without PTD. Among those with PTD, Met homozygotes scored ~2 SDs higher (20 points FrSBe total T-score), indicating substantially more behavioral problems than Val carriers. Similarly, among those with PTD, A2 homozygotes also reported scores that were ~2 SDs higher (20 points) than A1 carriers. Exploratory analyses suggested similar gene effects across all 3 FrSBe subscale scores, indicating that no single subscale was driving these results (data not shown). Multivariable analysis (see Table 2), controlling for premorbid psychiatric disorder, antidepressant use, and behavior before injury revealed a significant interaction between PTD status and COMT status at 12 months (n = 64; P = .007) but not at 6 months (n = 64; P = .427) postinjury. For ANKK1 status, this interaction was not significant at the 6-month (P = .553) time point. However, at the 12-month time point, there was a trend toward significance for an interaction between the ANKK1 variant (A2/A2) and PTD (P = .094). Compared with a base model without genetics or a PTD × Gene interaction (R2 = 0.436), adding a COMT × PTD interaction term accounted for 8% of observed variance (R2 = 0.516), whereas an ANKK1 × PTD interaction term accounted for 2.9% of observed variance (R2 = 0.465). For a preliminary analysis of potential cumulative/epistatic effects of these 2 genes, we conducted group analyses within our PTD population only at 12 months (n = 23). Assessment of individuals across both gene variants using ANOVA showed an overall effect of genotype grouping on behavior (F3 = 3.388; P = .039). Post hoc pairwise comparisons show that Met homozygotes/A2 homozygotes reported significantly worse behavior than Val carriers/A2 homozygotes (P = .044), and Val carriers/A1 carriers (P = .005); they also tended to perform worse than Met homozygotes/A1 carriers (P = .077); see Figure 2.
Multivariate Regression for Self-reported Behavior at 12 Months Postinjury: ANKK1 and COMT.
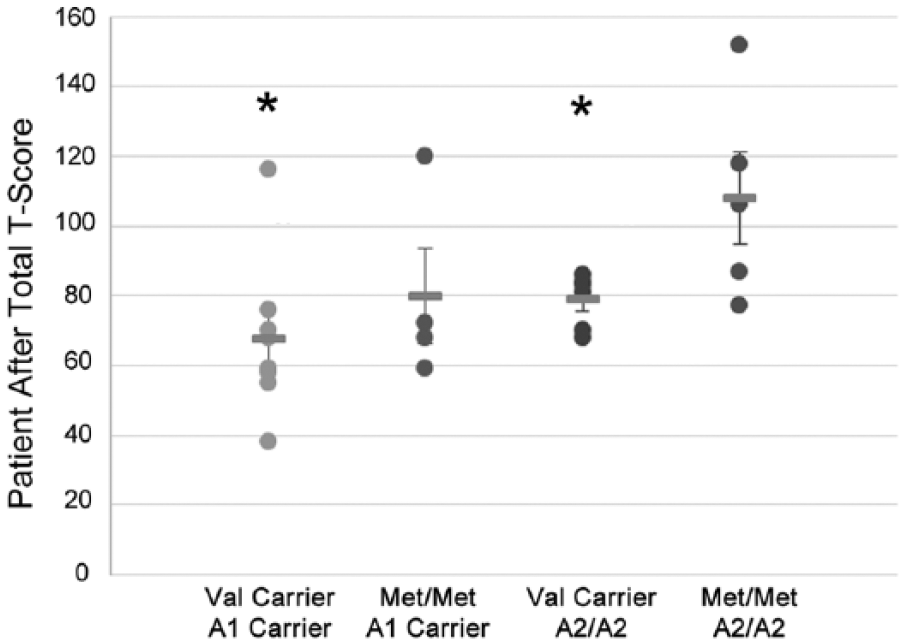
Individual FrSBe self-total after scores and group means are presented for Val158Met by ANKK1 groups in a PTD-only population (n = 23).
We conducted a secondary (bivariable) analysis using family-reported behavior measures to assess the impact of self-awareness on behavioral outcome measures at the 12-month time point (n = 57). The same group analyses were used as in the patient self-report (see Figure 1). A significant difference was once again observed between participants who were depressed and not depressed (P = .005). Depressed Met homozygotes performed significantly worse (~21 points) than their Val carrier counterparts (P = .040). No significant difference was observed between these genotypes in nondepressed populations (~7 points; P = .240). For Taq1A with PTD, there was no significant difference in family-reported behavior between depressed (P = .328) or nondepressed (P = .827) A2 homozygotes and A1 carriers.
Discussion
Taking a rehabilomics approach, this work represents the first study examining genetic associations with behavior post-TBI. We found no differences in behavior following TBI by Val158Met (COMT) or Taq1A (ANKK1) status alone; however, among those with PTD at 12 months, we found that Met homozygotes (Val158Met) and A2 homozygotes (Taq1A) self-reported significantly more behavioral problems 12 months post-TBI, supporting previous evidence that a relationship between DA genetics and behavior emerges in the context of a moderating stressor like PTD. Even when corrected for perceived preinjury behavior status, antidepressant use, and premorbid mood disorder, a significant interaction between Val158Met and PTD was still present. Because depression may disrupt PFC areas highly associated with effective and efficient top-down control of cognitive-behavioral processes,56,70 individuals with PTD may be especially susceptible to genetically mediated differences in DA system function. Importantly, we observed genetic associations only at 12 months postinjury. Because PTD develops most frequently during the first 6 months postinjury, 57 its effects on DA-moderated behavior may require time beyond 6-month PTD onset to emerge. Alternatively, depression may emerge following resolution of a chronically stressful state (eg, initial recovery from severe trauma). 56 Regardless, temporal relationships between mood and behavior might explain why we observed some trends at 6 months, with statistically significant results at 12 months postinjury.
Our results suggest that the severity of poor behaviors among those with PTD is related to Val158Met and Taq1A variation, and these findings are not simply attributable to PTD alone (Figure 2). Our hypothesis was supported, in that relative cortical DA system hyperfunction, presumably occurring among those homozygous for the COMT Met allele (high PFC DA levels), was associated with worse behavior when occurring with PTD. Furthermore, our hypothesis implicating the ANKK1 Taq1A variant in behavior within the context of PTD was also supported, although it did not hold up statistically in the multivariable model. We also provide preliminary evidence for an interaction between ANKK1 and COMT in the context of PTD-specific behavioral dysfunction. Although these findings need to be validated in an independent sample, the data demonstrate how this application of the rehabilomics framework 5 shows that DA genetics may contribute substantially to variability in behavior after TBI. Patients with identified DA genetic susceptibility could be monitored more closely for PTD development and managed with appropriate pharmaceutical, cognitive-behavioral, or other emerging therapies, and they could be provided earlier after injury to manage PTD and to prevent later development of severe behavioral symptoms.
Our previous work suggests that COMT Met homozygosity is associated with relatively better cognitive performance after TBI among women. 7 Though the disparate relationship between Val158Met and cognition versus behavior at first seems paradoxical, these findings support a growing body of evidence highlighting that a simple global hypodopaminergic/hyperdopaminergic model may not adequately describe complex cognitive-behavioral outcomes.61,71 These phenomena may be modeled better by considering regional alterations in DA system regulation that result in relative states of either hyperdopaminergia/hypodopaminergia. To this point, experimental TBI research suggests subcortical (striatal) DA deficits17,72 and increased medial PFC DA synthesis 73 following injury. These complex relationships may be further clarified when considering the tonic-phasic DA hypothesis, 21 as it applies to “PFC and striatal stability versus flexibility” as articulated by Bilder et al, 61 for both cognitive and behavioral function after TBI. Together with previous work evaluating DA genetics and cognition after TBI,7,47 this work represents the first clinical study supporting the tonic-phasic DA hypothesis.
The tonic-phasic theory of DA neurotransmission, as articulated by Grace,21(p1944) states that “the dynamics of DA regulation within limbic striatal regions occurs via two processes: (1) high-amplitude transient, phasic DA release mediated by DA neuron burst firing, and (2) constant low-level ‘background’ tonic DA that is regulated by baseline DA neuron firing and corticostriatal glutamatergic afferents.” Building on this framework, Bilder et al 61 suggest that within subcortical systems, high-amplitude phasic DA is released in conjunction with behaviorally driven bursts of action potentials. Phasic DA release is modulated by subcortical tonic DA levels via striatal presynaptic DA terminal D2 autoreceptors; glutamatergic corticostriatal afferents modulated by D1 receptor activity in the PFC also suppress phasic DA release. Because Met homozygosity likely increases tonic DA cortically and subcortically, these individuals may exhibit a relative suppression in mesostriatal DA neuron phasic burst firing, which could be further amplified by previously observed DA transporter reductions16,74 that limit DA clearance post-TBI. Interestingly, preliminary evidence suggests that experimental TBI reduces mesostriatal DA neuron phasic firing and production of spontaneous DA transients. 72 Striatal tonic/phasic DA modulation is distinct from DA actions in the PFC because of limited DA autoreceptor modulation, where Met homozygosity (increased DA) leads to more cortical excitability and more cortical-striatal inhibition of striatal phasic DA. 61 Work by Kobori et al 73 demonstrates that experimental TBI can increase PFC DA tone through D1 receptor mechanisms. Thus, genetic variations in DA pathways may accentuate disruption of the tonic-phasic interplay between cortical and subcortical DA systems associated with TBI.
In this context, the tonic-phasic DA model suggests that COMT activity could differentially influence top-down control (cortical-based cognitive-behavioral stability) and bottom-up stimulus-driven control of actions (subcortical cognitive-behavioral flexibility), which may be accentuated by depression. Clinically, after TBI, higher cortical DA levels associated with Met homozygosity may relatively preserve cortical DA system stability, important for neuropsychological performance, while simultaneously decreasing subcortical (phasic) DA transmission necessary for efficient changes in neural networks and flexibility in adapting to new situations and environments, important for functional cognition and navigating the real-world environment.75,76 Thus, the same DA levels that may be beneficial with neuropsychological test performance may lead to rigidity in thought and to fixation that are captured as relative deficits on behavior and functional cognition measures. Also, cortical suppression of subcortical phasic DA activity could further drive the depressive state56,70 that facilitated initial genetic associations with behavioral dysfunction after TBI.
D2 receptors primarily localized in the nucleus accumbens, caudate, and putamen also significantly affect behavior. A2 homozygotes tend to report more behavioral problems than A1 carriers in the context of depression, which in the setting of the cognitive-behavioral interplay associated with top-down control, is consistent with our previously reported Taq1A associations with cognitive outcomes. 47 One interpretation of our data is that D2 receptor density among A2 homozygotes with depression leads to difficulty in inhibiting maladaptive behaviors. Because D2 autoreceptor density can drive DA transporter expression, 77 the primary method for synaptic DA removal subcortically, 78 genetic variation within ANKK1, could also affect tonic DA availability subcortically after TBI. 74 With an injury-induced+stress-induced inability to modulate PFC function and its effect on bottom-up control, endogenous variations in striatal DA receptors may further blunt phasic responses and impair striatal flexibility to effectively manage behavior. 27 Our findings cannot elucidate the exact neurobiological mechanisms contributing to the apparent ANKK1 relationship with behavior, but the potential additive/epistatic effects of both Val158Met with PFC control of striatal DA systems and Taq1A-related subcortical DA system control may together facilitate poor behavior after TBI. Although this is an intriguing finding, our sample size for carriers of both risk alleles is small, and the particularly poor behaviors among those who were Met and A2 homozygotes with PTD need to be replicated in larger sample sizes.
These data suggest a hypothesis-generating theoretical rehabilomics-based framework for understanding the neurobiology of behavioral dysfunction in the context of a chronic stressor (PTD; Figure 3) that may inform mechanisms of neural recovery and pathways for personalized medicine approaches in neurorehabilitation. Individuals with relatively higher PFC DA activity and disrupted top-down PFC control associated with both TBI and PTD have an inability to activate appropriate subcortical DA system responsivity to respond flexibly to novel or changing conditions. With PTD, blunted cognitive flexibility and emotional control manifests as problematic behaviors. COMT Met homozygotes may have increased PFC DA levels and decreased subcortical phasic responses and, therefore, exhibit the proposed cortical response rigidity. A2 homozygotes with PTD have D2 function that magnifies the behavioral effects from decreased subcortical phasic DA responsivity that occurs in the context of COMT Met homozygosity. Thus, we propose that individuals who are both Met and A2 homozygotes with PTD would have both overactive PFC DA and decreased subcortical phasic DA responses that lead to greater susceptibility to dysregulated behavior. This hypothesis may help clarify what are seemingly contradictory findings with how the same DA genetic variants paradoxically preserve cognition yet facilitate poor behavior.
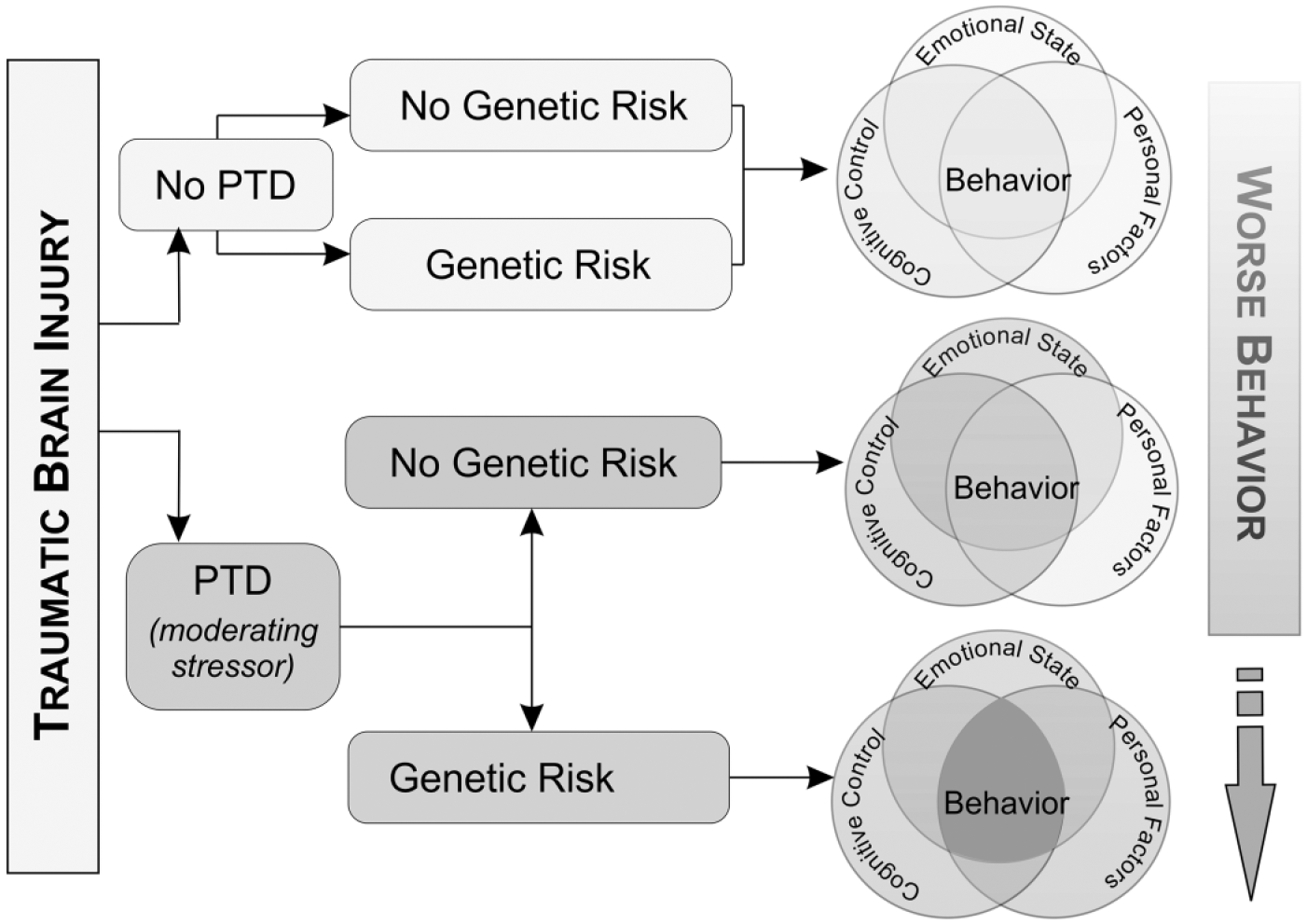
Behavior is a manifestation of cognitive control (cognition that can be measured with neuropsychological testing), emotional state (depression and anxiety), and personal factors (genetics or environment). Following TBI, an individual may exhibit deficits in none or all of these areas. For instance, individuals may have impairments in cognitive abilities but no underlying mood disorder and a favorable genetic profile. They are expected to exhibit few behavioral problems. Those with impaired cognitive abilities and a mood disorder but a favorable genetic profile may exhibit mild/moderate behavioral problems. However, those individuals with impaired cognitive control, a mood disorder, and an unfavorable genetic profile may exhibit the most severe behavioral problems. This work is just one example of how the rehabilomics framework can be used to better characterize patient outcomes.
A clear and integrated framework for how personal biology within DA systems encompasses both cognition and behavior may enhance existing strategies for TBI neurorehabilitation and repair. 79 Functional magnetic resonance imaging evidence suggests DA neuron dysfunction following TBI, with working memory activation related to Val158Met status. 80 This evidence of DA system dysfunction interacting with genetic susceptibility supports the notion that DA genetics inform clinical care. In rehabilitation, where strategies are needed to help individuals struggling with both chronic cognitive and behavioral disabilities, a rehabilomics framework would address both biological changes (through pharmaceutical therapies) and psychosocial changes (through behavioral interventions) to guide personalized-medicine approaches after TBI. With known pharmacological targets for both COMT 37 and D2 receptors 81 available, understanding how to target DA systems based on individual genetics and symptom (cognitive vs behavioral) profiles may provide more precision with effectively managing these individuals pharmacologically. Studies already show differential responses to antidepressant treatment by Val158Met status.82,83 Furthermore, a study in populations with schizophrenia showed that Met carriers may be more responsive to deficit-targeted computerized cognitive exercises to improve quality of life. 84 Future work may focus on using these same markers in determining which patients may benefit the most from cognitive rehabilitation and pharmaceutical therapies after TBI.
The population with severe TBI is a highly specialized rehabilitation population, with a relatively low incidence compared with other clinical populations. Although our sample is small, compared with large population-based genetics studies, and validation in larger and more ethnically diverse samples is needed, numerous aspects of this study support the veracity of the findings. The literature strongly supports a functional role for the selected genetic polymorphisms; thus, we examined these known functional variants in the context of a new clinical population (TBI). Additionally, we demonstrate the potential clinical relevance of these functional polymorphisms in TBI, with significant associations in cognitive7,47 and behavioral performance in this sample at the same assessment time points.
Because our previous studies suggest that sex influences genetic relationships with cognitive outcomes, future work should investigate if sex moderates relationships between the risk groups identified in this study and behavior. Investigation of temporal relationships between PTD onset and appearance of behavioral symptoms, particularly in response to acute stressors, within the context of DA genetics is also needed. Nonetheless, our work provides initial evidence that clinical studies examining DA system functional relationships to cognitive outcomes and behavior may identify appropriate and personalized management strategies more effectively than what is currently available, and these findings represent a robust early example of how rehabilomics-based research applications may lead to personalized approaches to TBI care.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: National Institute For Disability, Independent Living, and Rehabilitation Research (NIDILRR): 90DP0041, National Institutes of Health (NIH): R01 HD048162; Department of Defense (DoD): W81XWH-071-0701.