
Abstract
Neurogenesis, the birth of new neurons, occurs throughout life in the subventricular zone and produces immature neurons that migrate tangentially through the rostral migratory stream to the olfactory bulb. This migration is tightly regulated by both structural and chemical influences. Interestingly, brain insults such as ischemic stroke increase neurogenesis and redirect neuroblast migration to the injury site. This injury-redirected neurogenesis and migration is coupled with angiogenic vasculature and is influenced by many of the factors that positively and negatively affect migration under developmental or normal adult conditions. Additionally, cytokines and chemokines such as stromal cell-derived factor-1 strongly influence neuronal migration poststroke. However, neuronal repopulation or brain regeneration is extremely limited. This limitation may potentially be due to the hostile poststroke microenvironment including the formation of the physical and chemical barriers of glial scar. Furthermore, interspecies differences in poststroke neurogenesis between rodents and humans complicate the translation of experimental results to humans. Despite these challenges, many drugs and other potential therapies have recently been evaluated for potential neurogenic properties poststroke. Improved understanding of poststroke neurorepair may lead to new and more effective neurorestorative therapies.
Physiological Adult Neurogenesis and Migration Through the Rostral Migratory Stream
Neurogenesis, the birth of new neurons, occurs throughout life in the subgranular zone (SGZ) of the hippocampal dentate gyrus and in the subventricular zone (SVZ) lining the lateral ventricle. In addition to radial migration of neuroblasts from the SVZ to the cortical surface during development, neuroblasts from the adult SVZ migrate tangentially through the rostral migratory stream (RMS) to the olfactory bulb (OB). In this process, “homophilic” migration describes how neurons facilitate each other’s migration in chains, resulting in saltatory movement through the RMS. Additionally, astrocyte endfeet influence migration by surrounding the RMS and creating a nearly encased tube. Finally, blood vessels profoundly influence neuronal migration by releasing protective factors and acting as a physical scaffold, thereby providing a path of least resistance for migrating neuroblasts. Neuronal migration is influenced by cell-secreted factors and by cell-bound molecules including GABA, VEGF, BDNF, PSA-NCAM, matrix metalloproteinases, L1 CAMs, β1 integrins, netrins, slits, ephrins, semaphorins, and extracellular matrix (ECM) components (see Kahle and Bix 1 for review and Table 1 for abbreviation definitions). Interestingly, neurogenesis is increased after brain injury and may be a component of brain self-repair and regeneration. This review will focus on the processes, influences, effectiveness, and barriers of endogenous neurorepopulation following ischemic stroke. The importance of and current investigations in experimental neuroregenerative treatments will also be discussed.
Abbreviation Definitions (Order of Appearance).
Neuroblast Migration Following Ischemic Stroke
Following transient middle cerebral artery occlusion (MCAO) models of ischemic stroke, the ipsilateral SVZ is transiently hypoxic as detected by hypoxyprobe-1 immunohistochemical staining. 2 Stroke increases cell proliferation within both the SGZ3,4 and SVZ,5-8 and cells attempt to migrate toward the infarct area rather than the OB (as evidenced by BrdU in the vast majority of studies; DiI, tritiated thymidine, PCNA, or Ki67 are sometimes used for new cell labeling and tracking), while differentiating into neurons and astrocytes. Although SVZ neurogenesis poststroke is the focus of this review, SGZ neurogenesis also occurs under normal, stroke, and other central nervous system injury conditions (reviewed by Hodge and Hevner, 9 Masiulis et al, 10 and Cho and Kim 11 ). Furthermore, neural stem cells may be present and undergo neurogenesis in other brain regions, including the hypothalamus (reviewed by Migaud et al 12 ), cortex, striatum, and amygdala (reviewed by Kokaia and Lindvall 13 ). However, some contradictory studies suggest that neurogenesis in these regions may only be the result of neurogenesis originating in the SVZ or SGZ (reviewed by Landgren and Curtis 14 ).
Immature, migrating neuroblasts can be stained and quantified by their expression of doublecortin (DCX), a microtubule-associated protein that appears to play a role in organization and stabilization of microtubules.15-17 Increased neurogenesis and migration begin within the first week, persist for several weeks, and possibly return to baseline at 6 weeks following stroke injury in the rodent.2,5,8,18,19 However, in the adult rat, increased neurogenesis may persist as long as 4 months after stroke. 20 Neurogenesis within the SVZ following stroke is stimulated by many of the same signals discussed previously for normal conditions. 1 Nitric oxide donors also increase neurogenesis in the ipsilateral SVZ following ischemic injury and improve functional outcome. 5 Additionally, retinoic acid, a vitamin A derivative, and chemokines also increase poststroke neurogenesis. 21
Cells migrating from the SVZ after stroke exhibit many of the same characteristics of those migrating under nonpathologic conditions, including forming elongated chain aggregates and associating with astrocytes and blood vessels. 22 However, these cells are presumably redirected from their typical RMS–OB pathway toward the site of injury in an endogenous attempt at regeneration and neuronal repopulation (self-repair) of the lesion area.6,18,23 This redirection could be mediated by disruption of the mechanisms that typically maintain neuroblasts along their route through the RMS to the OB and promotion of mechanisms that enhance migration toward the injury site. 22
Angiogenesis–Neurogenesis Coupling Poststroke
As under normal conditions, SVZ neurogenesis, neuroblast survival, and migration from the SVZ following ischemic stroke are promoted by the vasculature and coupled with the process of angiogenesis. Many pro-angiogenic genes such as VEGF are upregulated as soon as 1 hour after ischemia, and some pro-angiogenic genes such as angiopoietins 1 and 2 are upregulated even 21 days after ischemia. 24 Furthermore, angiopoietin-2 is upregulated specifically within the SVZ following stroke and influences both the migration and differentiation of neuroblasts. 25 Endothelial cell proliferation and blood vessel number often increase as soon as 1 day and 3 days after injury, respectively, and persist for at least 21 days after injury in rodent experimental models. 24 Interestingly, neuroblasts born after stroke closely associate with and interdigitate around the vasculature as they migrate toward the site of injury and can be found within the peri-infarct area where angiogenesis is induced after stroke.2,23,26 VEGF is an important factor in this poststroke angiogenesis–neurogenesis coupling. VEGF administration via cannula starting 24 hours poststroke in rodents results in more BrdU (bromodeoxyuridine, a synthetic nucleoside incorporated into DNA that is used as a label and tracker for newly born cells) colocalization with neuronal lineage markers in the SVZ as compared to controls. 27
Stromal Cell–Derived Factor-1 Increases Neuronal Migration and Homing to the Stroke Site
In addition to the mechanisms by which the vasculature acts as a migration scaffold and source of proneurogenic and migratory factors, cytokines and chemokines, some released by the vasculature, also heavily influence the rate and guidance of neuroblast migration following stroke. Stromal cell–derived factor 1 (SDF-1) and its receptor CXCR4, expressed on migrating neuroblasts, are especially important.20,23 In in vitro and in vivo rodent hypoxic-ischemic cerebral injury studies, SDF-1 enhances neurogenesis and promotes chemoattractive chain migration and transmigration in a CXCR4-dependent manner. 28 Following experimental focal cerebral ischemia, SDF-1 is upregulated in blood vessels of the penumbra (ischemic at-risk area surrounding the necrotic core) and decreased in nonlesioned brain areas including the contralateral hemisphere. 29 This expression pattern suggests that SDF-1 may be both encouraging migration toward the area of injury and discouraging migration toward other areas. Hypoxia inducible factor (HIF)-1 may be at least partly responsible for the upregulation of SDF-1 in endothelial cells at sites of ischemic injury. 29 Interestingly, in addition to the SDF1-β isoform expressed in cerebral microvascular endothelial cells, the alternatively spliced SDF1-α isoform expressed in neurons is also preferentially upregulated at the site of focal cerebral ischemia. Therefore, by secreting SDF1-α, distressed peri-infarct neurons may call migrating neuroblasts for help (secretion of beneficial factors) or to replace them if they die (regeneration). Specifically, SDF-1 promotes the migration and homing of CXCR4-expressing cells into the site of injury.20,28-32 Accordingly, blocking CXCR4 following MCAO inhibits migration of neuroblasts out of the SVZ. 20 Interestingly, intracerebral administration of SDF-1 following experimental cerebral ischemia induced the homing of bone marrow–derived cells from peripheral blood to the damaged brain and specific site of injury. This resulted in decreased infarct volume due to neuroprotection and CXCR4-expressing cell repopulation of neurons (BrdU-positive colocalization with Neu-N and MAP-2) and astrocytes (BrdU-positive colocalization with GFAP) at the site of injury. Furthermore, SDF-1 treatment also enhanced poststroke angiogenesis in the ipsilateral hemisphere. 31 Also, animals had a drastic increase in BrdU+ cells when an adenovirus expressing the SDF1-α gene was intracerebrally injected into the boundary of the infarct area. This suggests that delayed (1 week) poststroke administration of SDF1-α increased neurogenesis, migration, and differentiation, which was effective for long-term recovery and distinct from neuroprotective effects. 33
SDF-1 Drives a Positive Feedback Loop of Angiogenesis and Neuronal Migration
As SDF-1 directly promotes angiogenesis, a positive feedback loop may exist whereby SDF-1 is both produced by and promotes the formation of the vasculature. Indeed, SDF-1 increases endothelial cell proliferation and, potentially via a PI3K/Akt pathway, expression of VEGF.34,35 Interestingly, domain V (DV), a protein fragment of the extracellular matrix component perlecan that we have identified as a potential novel stroke therapy, also increases angiogenesis in the brain and stimulates VEGF production and release from brain endothelial cells via a PI3K/Akt pathway.36,37 Furthermore, local SDF-1 can augment neovascularization of ischemic tissues by attracting endothelial progenitor cells, which also express CXCR4, thereby inducing subsequent angiogenesis and vasculogenesis via a VEGF/eNOS-related pathway.38,39 Finally, SDF-1 and angiopoietin-1 appear to causally link neurogenesis with angiogenesis within the peri-infarct neurovascular niche after ischemic stroke. 26 Collectively, the vasculature at the site of stroke injury increases SDF-1, which directly acts as a homing signal and promigratory factor for neuroblasts, upregulates factors such as BDNF to indirectly increase migration efficacy, and increases angiogenesis. Angiogenesis then creates new vascular scaffolds for neuroblast migration toward the site of injury and provides a larger source of SDF-1 secretion. 2 These positive feedback mechanisms are depicted in Figure 1.
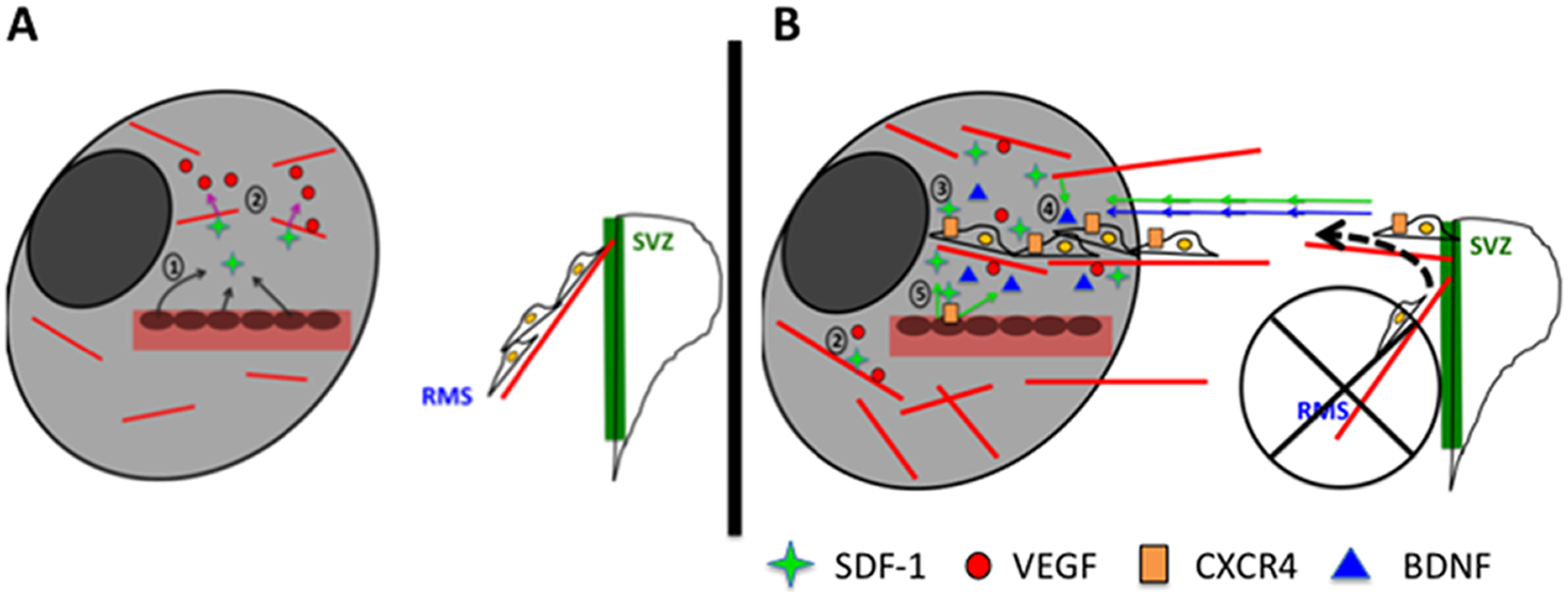
SDF-1 facilitates both angiogenesis in and neuroblast migration to the ischemic stroke site in a potential positive feedback loop. (a) Cartoon depiction displaying how, following ischemic stroke, the 1) expression of stromal cell-derived factor (SDF)-1 is increased in peri-infarct (light gray) regions, and 2) SDF-1 is proangiogenic and supports the formation of new vasculature by mechanisms including increased VEGF expression, while (b) 3) SDF-1 directly acts as a promigratory cytokine to migrating neurons that express its CXCR4 receptor while also 4) inducing the release of BDNF, which also serves as a promigratory cytokine for neuroblasts, and 5) by forming new vasculature, SDF-1 may provide a larger source for more of its production, the production of other protective factors, and more vascular scaffolding for neuroblasts to migrate upon.
While this strong connection between SDF-1 and vascularization exists, the endothelium is not the sole source of production of SDF-1 in the infarcted and ischemic areas. Indeed, astrocytes and microglia are also considered to be important sources of the chemokine at the site of insult.20,26,28 Therefore, within the hostile microenvironment of the ischemic stroke infarct and surrounding ischemic areas there appears to be a collaborative effort to increase SDF-1 production to attract neuroblasts to the area of damage for potential cell repopulation.
Other Factors Involved in Poststroke Neurogenesis and Migration
Additionally, other promigratory or proneurogenic cytokines and chemokines are upregulated at the site of rodent stroke injury in astrocytes and microglia. These include CCL2 (also known as monocyte chemoattractant protein-1, MCP-1), which attracts CCR2- and CCR5-expressing SVZ neuroblasts toward the ischemic lesion,40,41 VEGF, EGF (epidermal growth factor), granulocyte-macrophage colony-stimulating factor (GM-CSF), BDNF, and bFGF. 26 Ephrin B2 is also upregulated following ischemic stroke in the peri-infarct area where it influences neuroblast proliferation and migration, in addition to influencing vascular sprouting and mural cell recruitment for maturing blood vessels, and angiogenic growth factors upregulate ephrin B2 in turn.26,42
Matrix metalloproteinases (MMPs) 2 and 9 are also strongly upregulated for several weeks after stroke, and reduction of MMP activity with the broad-spectrum inhibitor GM6001 drastically decreases the amount of migrating BrdU+/DCX+ cells reaching the damaged area. While MMPs aid in the normal migration of neuroblasts, they are also believed to be especially important following injury in helping cells degrade their way through the ECM as they migrate to the damaged area.26,43,44 Similarly, cathepsins, another class of proteases, are differentially modulated and play a role following stroke. For example, Cathepsin B is upregulated in vitro in response to OGD (oxygen glucose deprivation, a model of stroke) and increases the processing and release of perlecan’s C-terminal portion of DV, LG3. LG3, in turn, is neuroprotective against OGD in rodent primary cultured neurons.45,46 The ability of DV and LG3 to increase neuronal repopulation following experimental rodent stroke is an active area of investigation in our lab.
Difficulty of Newborn Neurons to Reach and Repopulate the Infarcted and Ischemic Areas: Role of the Glial Scar
The brain attempts to repair itself after stroke via a variety of mechanisms to increase neurogenesis and neuroblast migration toward the site of stroke damage. However, despite several weeks of enhanced neurogenesis and migration, few neuroblasts are successful at reaching, maturing, and repopulating the damaged area. This is most likely due to the hostile poststroke environment.5,6,23,26 In the acute phase following stroke, cortical ischemia itself impedes neuroblast travel. 47 Furthermore, the numbers of newborn neurons that do repopulate the stroke area are too small for recovery of neurological function. 6 Additionally, successfully migrated neurons would need to extend neurites and connect to other neurons to effectively regenerate tissue. One major facet complicating these events for new neurons is the glial scar formed after injury.
Most of our understanding of glial scar and its effects on neurons derives from spinal cord studies. These studies have demonstrated that glial scar is formed primarily by hypertrophic reactive astrocytes and is largely composed of chondroitin sulfate proteoglycans (CSPGs) and keratan SPGs. Although the glial scar is beneficial in the acute phase of injury to help quarantine injured tissue, prevent a hyperinflammatory response, and repair the blood–brain barrier, it becomes a physical and chemical detriment to repair in the chronic phase.48,49 For example, Semaphorin 3, 50 Ephrin-B2, 51 and Slits 52 are upregulated and associated with glial scar following various central nervous system injuries, and each of these factors has been identified as a chemorepulsive cue to immature migrating neurons in rodents (see Kahle and Bix 1 for further review). Although it is not known whether each of these translates to the glial scar generated specifically after ischemic stroke, this is a logical hypothesis. Indeed, ephrin-A5 is released from reactive astrocytes in the peri-infarct cortex and inhibits both axonal sprouting and motor recovery in mice. 53 Additionally, the glial scar leads to inhibition of oligodendrocyte precursor cell migration and recruits inflammatory and other cell types that secrete factors, such as tenascin-R, 54 that inhibit axon regeneration. Tenascin-R stops and dissociates migrating neuron chains (eg, at the OB) in rodents. 55
Further complicating the scenario, different types of neurons may have differing susceptibilities to the inhibitory effects of CSPGs. 48 CSPGs, in turn, may be able to convert chemoattractive cues to chemorepulsive cues. For example, thrombospondin repeats within Semaphorin 5A may interact with the glycosaminoglycan portions on either heparan or chondroitin SPGs resulting in axonal attraction or repulsion, respectively. 56 Given the potentially long distance and time of axonal remodeling following stroke, especially in humans, these findings could have broad implications for how much the glial scar could influence the poststroke microenvironment. Thus, the glial scar could potentially inhibit neuronal repopulation by multiple mechanisms, including (a) physically blocking the neuronal migration (or axon extension) path and access to promigratory cues, (b) chemically repelling or slowing migrating neurons, and (c) converting promigratory cues into inhibitory cues.
Unsuccessful axon extension/regeneration in the glial scar environment results in the formation of classically termed axonal “dystrophic endings,” which have dynamic but not successful repairing activity. 48 Chondroitinase ABC, an enzyme that degrades CSPGs, reverses scarring and axonal inhibitory effects of CSPGs in vitro and in vivo. When infused directly into stroke infarct cavities, beginning 7 days after rat MCAO, chondroitinase ABC reduced GFAP reactivity, increased peri-infarct MAP-2 reactivity, improved motor function, and increased neurite extension in cortical neuron cultures (associated with increased BDNF). 57 These important studies suggest that reducing the glial scar and/or increasing neurite extension, distinct from poststroke neuroprotection (given the delayed initiation of treatment after stroke), are viable treatment options for ischemic stroke. Importantly, although much of the work characterizing the glial scar focuses on axon extension, many of the influences (eg, chemoattractive and chemorepellent cues) on axon extension also translate to neuronal migration, and both processes are necessary for neuronal repopulation/regeneration poststroke.
Implications for Human Stroke
Many relevant differences exist between the human and rodent brain (typically used in stroke experimentation) that influence the role and therapeutic potential of poststroke neurogenesis. For example, the human RMS may be active only at very young ages and may target the prefrontal cortex and OB. 58 The architecture and organization of stem cells within the human SVZ differs from the rodent and is characterized by an astrocyte ribbon separated from the ependyma by a hypocellular gap layer. Additionally, immature neurons emerging from the human SVZ do not migrate in chains.1,59 Human neuroblasts also have a significantly longer distance to migrate to the damaged area (and axons have farther to extend) compared with animal models with much smaller brains. 26 Finally, evidence from nonhuman primates suggests that the time window for neurogenesis and migration may be considerably longer compared with rodents. Indeed, one study showed that 3 cynomolgus monkeys had DCX-positive migrating neurons in the striatum close to the SVZ, although only 1 monkey had robust migration at 4 weeks following a photothrombotic stroke. 60 Also, the time window from neurogenesis in the SGZ to maturation of granule cells may be more than 6 times longer in macaques (6 months) than in rodents. 61 Prolonged neuroregeneration may provide a longer and more accessible treatment window in humans. This far from comprehensive list reminds us that poststroke neurogenesis differences exist between species and that rodent stroke models may not translate well to human stroke. However, the impacts of most interspecies differences on translation are not currently known.
Interestingly, increased proliferation has been observed in the human ipsilateral SVZ (as compared with the contraleral SVZ) following ischemic stroke as evidenced by an increase in the width and cell density of the astrocyte ribbon and gap layers, an increase in Ki-67+ cells, and an increase in Tuj-1+ and PSA-NCAM+ cells (neuronal and migrating cell markers). 62 In other human studies, Ki67+ cells expressing DCX, βIII-tubulin, and/or TUC-4 were localized to the ischemic penumbra and a presumed neurovascular niche was identified where DCX+ cells were clustered near von Willebrand factor+ vasculature and Nestin+ cells. 11 Increases in Nestin+ cells and Musashi-1+ cells, potentially neural stem/progenitor cells, were also found in the infarct border area of a human brain at autopsy following cardiogenic cerebral embolism. 61 In other studies, recovering stroke patients over the age of 60 years were found to still possess an increase (although not as robust as in younger individuals) in Ki67+ cells and DCX+ cells in the SVZ, without apoptosis. 9
Therapies Needed to Enhance Neuronal Repopulation of Stroke-Affected Areas
Importantly, decreasing neurogenesis, either by genetic ablation of DCX in mice63,64 or by focal irradiation of the SVZ in gerbils, 65 worsens outcomes following experimental stroke and ischemia. On the other hand, transgenic mice overexpressing VEGF, or rats given VEGF-expressive plasmids into the lateral ventricle, show increased poststroke neurogenesis, migration, and repopulation of BrdU+/Tuj1+, BrdU+/MAP-2+, and BrdU+/GAD67+ neurons as well as increased neurite length and branch numbers. Furthermore, these increases were associated with improved poststroke outcomes.66,67
Therefore, although barriers exist, several experimental ischemic stroke therapies exploit the poststroke reparative and regenerative response. Additionally, several successful treatments that were developed based on a different rationale have subsequently been found to enhance neuroregeneration. For example, citicoline, an endogenous compound currently in clinical phase III trials for acute ischemic stroke, has been supported for its neuroprotective potential; however, it also increases neurogenesis in the SGZ, SVZ, and the peri-infarct area. These results were accompanied by improved sensorimotor function and increased NMDAR- and AMPAR-binding densities (signs of an excitation shift) in the perilesional cortex. 68 Additionally, intraventricular administration of EGF and albumin increases neuronal replacement 100-fold in stroked mice. 69 Also, cilostazol, a type 3 phosphodiesterase inhibitor, reduces infarct volume and time for recovery of neurological deficit as well as enhances the number of BrdU+ and BrdU+/DCX+ cells in the ipsilateral SVZ and peri-infarct areas following transient focal cerebral ischemia in mice. 70
Importantly, SVZ-derived neuroblasts are not only capable of arriving at the damaged site but also fully differentiating into neurons, extending processes, and forming synapses with neighboring cells. 22 This repopulation concept is depicted in Figure 2. Indeed, valproic acid, a pan histone deacetylase (HDAC) inhibitor (typically used to treat seizures), given beginning 24 hours after MCAO, increases neurogenesis at the SVZ, neurite length, glutamate transporter 1 expression (which colocalized with synaptophysin at the ischemic boundary), and improves motor function 28 days poststroke in rats. 71 Neurogenic, promigratory, and differentiation (to expression of NeuN) effects following stroke were also found following administration of sodium butyrate, another HDAC inhibitor. These beneficial effects depended on BDNF-TrkB signaling and downstream activation of CREB. 72 Likewise, LM22A-4, a small molecule designed to mimic BDNF agonist activity on TrkB, improved motor function and increased neurogenesis/ neuronal repopulation when treatment was initiated 3 days poststroke, suggesting an important role for TrkB in poststroke neurogenesis in mice. 73
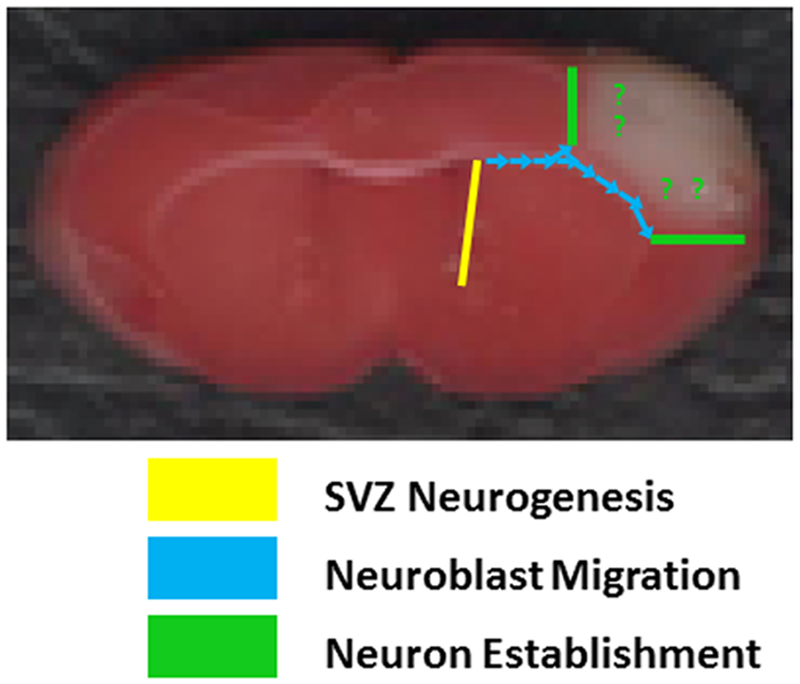
Neuronal migration following experimental cortical stroke.
Postischemic neurogenesis, migration, and differentiation also occurred in rats where SGZ cells chronologically expressed BrdU, the transcription factor NeuroD, DCX, NeuN, and MAP-2; repopulated the hippocampus; and responded to NMDA stimulation. 74 Interestingly, new neurons repopulating the damaged area following stroke preferentially differentiate into the phenotype of the neurons destroyed by the injury.6,7 Although this is likely in response to site-context-specific cues, the underlying mechanisms are still unknown (to the best of our knowledge). Indeed, following global ischemia, intraventricular infusion of EGF and bFGF resulted in massive regeneration of the damaged CA1 region (by cells born in both the SGZ and SVZ) with cells that morphologically and electrophysiologically appeared to be pyramidal neurons and persisted for at least 6 months. Additionally, these animals had improved recovery of spatial learning deficits in the Morris water-maze, providing further support for the importance of enhancing endogenous neurogenesis, migration, and repopulation for restoration of function. 75 Interestingly, salvianolic acid B, a traditional Chinese herbal medicine used for clinical stroke in China, boosted neurogenesis and improved cognitive impairment in the Morris water-maze with a delayed, poststroke day 7, treatment initiation. 76 This suggests that these specific effects were not due to neuroprotection and that targeting neurogenesis as a stroke therapy can be effective with delayed treatment. Other molecules being investigated for their ability to enhance the endogenous neurogenic response following ischemic stroke or ischemia include meteorin, 77 leptin, 78 melanocortins (investigated in the hippocampus), 79 estrogen, 80 and others.
These steps of endogenous regeneration—neurogenesis, migration, repopulation, differentiation, neurite extension, and synaptic connectivity and integration—are essential for functional neuronal repopulation of the damaged area, and the findings that the brain is capable of this feat is a marvel of self-repair and encouraging for ischemic stroke therapeutics.
Additional Considerations for Potential Poststroke Neurogenic Therapy
New neurons that migrate to the sites of stroke injury may provide benefits independent of neuronal replacement. For example, new neurons, as seen in some stem cell therapies, may secrete beneficial factors. Thus, endogenous progenitor cells could potentially act as vehicles that bring a source of neurotrophic support to the remaining cellular elements at the site of injury. For example, neural progenitor cells of the mouse neonatal SVZ secrete neuroprotective sonic hedgehog in culture. 81 Some experimental stroke studies that suggest a neurogenic component to their treatment mechanism, for example, VEGF, concede that improvements occurred too soon after ischemia for mature neuronal replacement to be responsible. 27 On the other hand, ablation of neural progenitors also causes deficits too early to be accounted for by lack of mature neuronal replacement and may be the result of lack of chemical mediators. 63 Therefore, time after injury is one aspect that may help discern the type of progenitor benefits. Studies designed to test the progenitor benefits of neuronal replacement rather than solely neurotrophic support might consider delaying initial treatment, for example, 7 days after injury, and also tracking the progenitors to determine if they survive and integrate into networks.
Therefore, enhancing neurogenesis/migration after stroke may be beneficial even if the new repopulating neurons do not survive long term. This consideration is especially important given the controversial nature of neurogenic and repopulating therapies within the stroke field. Indeed, the concept of potentially enhancing neurogenesis, migration, and repopulation/regeneration is similar to the rationales of exogenous stem cell administration. One key difference, however, is that this approach may avoid several potential limitations by facilitating endogenous stem cells rather than administering exogenous ones. For example, when (how long after stroke), where, and how (route of administration) to optimally deliver stem cells, in terms of efficacy and minimizing side effects, is still far from being determined. 82 On the other hand, manipulation of endogenous neurogenesis might face similar issues of targeting even once an understanding of the endogenous neurogenic time course is more established. This is especially valid for the translation of potential stroke treatments, as the neurogenic architecture and process following stroke may, again, be very different spatially and temporally between rodents and humans. 58 Future studies will need to address the optimal species- specific method to target endogenous neurogenesis, which could potentially be weeks or months after injury, and how to couple interventions with rehabilitation and exercise-related neurogenesis. 83
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.