
Abstract
Gingival overgrowth occurs mainly as a result of certain anti-seizure, immunosuppressive, or antihypertensive drug therapies. Excess gingival tissues impede oral function and are disfiguring. Effective oral hygiene is compromised in the presence of gingival overgrowth, and it is now recognized that this may have negative implications for the systemic health of affected patients. Recent studies indicate that cytokine balances are abnormal in drug-induced forms of gingival overgrowth. Data supporting molecular and cellular characteristics that distinguish different forms of gingival overgrowth are summarized, and aspects of gingival fibroblast extracellular matrix metabolism that are unique to gingival tissues and cells are reviewed. Abnormal cytokine balances derived principally from lymphocytes and macrophages, and unique aspects of gingival extracellular matrix metabolism, are elements of a working model presented to facilitate our gaining a better understanding of mechanisms and of the tissue specificity of gingival overgrowth.
Introduction
Clinically detectable fibrotic overgrowth of gingiva is caused by a variety of etiological factors and is exacerbated by local bacterial plaque accumulation. Gingival overgrowth can be inherited (hereditary gingival fibromatosis), or is of idiopathic origin, or is sometimes associated with other systemic diseases. The majority of cases, however, occur as a side-effect of systemic medications. These medications include the anti-seizure drug phenytoin, the immune suppressor cyclosporin A, and certain anti-hypertensive dihydropyridine calcium-channel-blockers, most notably nifedipine. Clinical characteristics of different forms of gingival overgrowth have been previously reviewed (Hassell and Hefti, 1991; Marshall and Bartold, 1999; Seymour et al., 2000). There is now general agreement that gingival overgrowth lesions all contain fibrotic or expanded connective tissues with various levels of inflammation and an enlarged gingival epithelium. The degrees of inflammation, fibrosis, and cellularity depend on the duration, dose, and identity of the drug, on the quality of oral hygiene, and on individual susceptibility that stems from genetic factors and environmental influences. The present review summarizes the importance of gingival overgrowth as a clinical problem, and highlights recent advances in the identification of molecular and cellular processes that are leading to an enhanced understanding of the biological mechanisms underlying the overgrowth. Recent genetic studies on hereditary gingival fibromatosis are reviewed. These studies are beginning to identify the unique cell biology and extracellular matrix metabolism that occur in human gingiva, and a working model is presented. The term ‘gingival overgrowth’ will be used throughout the current text so that the biological aspects of this heterogeneous clinical condition will not be limited to hyperplasia or hypertrophy.
Clinical Importance of Drug-induced Gingival Overgrowth
Drug-induced gingival overgrowth is a side-effect and unwanted outcome of systemic medication and is limited to gingival tissues. Drug-induced gingival overgrowth does not originate in the periodontium, but it occurs exclusively in periodontal tissues and is generally not of the same magnitude in other tissues. Patients who receive anti-epileptic therapy through the systemic use of phenytoin (diphenylhydantoin or Dilantin®), or immunosuppressive therapy with systemic cyclosporin A (Sandimmun®), or anti-hypertensive treatment with systemic nifedipine develop gingival overgrowth with various degrees of prevalence. While the incidence of this side-effect can be as high as 65% in epileptics, 70% in transplant patients, and 30% in hypertension subjects, variation exists in the reported prevalence and severity of the clinical problem (Hassell, 1981; Lucas et al., 1985; McGaw et al., 1987; Barclay et al., 1992; Hancock and Swan, 1992; Thomason et al., 1995, 1996). Clinical and histological characteristics of drug-induced gingival overgrowth include hyperplasia in junctional epithelium and hypertrophy in keratinized epithelium (Ayanoglou and Lesty, 1999) and excessive connective tissue accumulation. In addition to being disfiguring and uncomfortable for affected individuals, moderate to severe forms of gingival overgrowth impair oral hygiene and may lead to increased accumulation of micro-organisms. Oral infections can potentially impair systemic health (Casamassimo, 2000; Li et al., 2000), and elevated oral infections stemming from gingival overgrowth could possibly compromise the general health of patients. This is most obvious in the case of organ transplant recipients who require continuous therapy with cyclosporin A, thereby being rendered critically susceptible to life-threatening systemic infections, and who concomitantly develop gingival overgrowth. The effective management of these patients clearly requires the active involvement of both dental and medical professionals to minimize the possibility of complications.
Dose-dependent correlations with the severity of gingival overgrowth are weak, but decreased drug use in general results in reduced severity of gingival pathology. For example, phenytoin was reported to effuse into crevicular fluid without any correlation to the incidence of overgrowth (McLaughlin et al., 1995), while no direct link was shown between overgrowth and the concentrations of phenytoin and metabolites (Ball et al., 1996). A more recent study supports a correlation between diminished metabolism of phenytoin in affected individuals and overgrowth (Kamali et al., 1999), but this has not been confirmed. Age, gender, concomitant medication with multiple drugs, local factors such as plaque accumulation, and genetic disposition are additional complicating risk factors in drug-induced gingival overgrowth (Thomason et al., 1995, 1996; Cebeci et al., 1996).
Therapy of drug-induced gingival overgrowth would seem to be most simply accomplished by the use of alternate medications that do not induce gingival overgrowth, and new medications are under development. At this time, however, older medications are still in active use. Cyclosporin A, for example, increases the rates of survival of most organ transplant patients, although the more recently developed immunosuppressant FK506 (tacrolimus) can be substituted in some patients (Bader et al., 1998; Busque et al., 1998; Kohnle et al., 1999; Thorp et al., 2000). Tacrolimus has not yet been associated with gingival overgrowth, but does cause other side-effects that are important for some patients (Thorp et al., 2000). The literature on the effective substitution of cyclosporin A with tacrolimus is relatively new. Cyclosporin A continues to be the most commonly prescribed drug for the prevention of graft rejection. Similarly, although several new anti-seizure drugs have been introduced and its prescription is markedly reduced, phenytoin remains the drug of choice for certain types of grand mal epileptic seizures (Wilder, 1996; Hupp, 2001). Finally, although hypertension cases are now being treated by alternative calcium-channel-blockers that either do not predictably cause gingival overgrowth (Westbrook et al., 1997; Ellis et al., 1999) or are linked to isolated cases of gingival pathologies, nifedipine remains a highly effective agent in the management of patients who do not respond sufficiently well to other anti-hypertensive medications (Messerli et al., 2000; Midtvedt et al., 2001; Leenen et al., 2002). In summary, it should be anticipated that drug-induced gingival overgrowth will continue to be a problem until safe and equally reliable medications to control these systemic conditions are developed and fully utilized. In addition, because the administration of these medications in general is beyond the control of the dental professional, clinical management of the gingival overgrowth presents a continuous challenge.
Treatment of the gingival overgrowth lesion itself can be complicated due to the superimposed inflammation on the fibrotic tissue enlargement. Traditionally, periodontal therapy offers removal of the inflammatory component of the overgrowth through scaling and gingival curettage, followed by excision of the overgrown gingiva (Kimball, 1939; Hassell and Hefti, 1991). For patients with severe gingival overgrowth and who require continuous drug therapy for medical reasons, gingivectomy must be repeated periodically due to the recurrent nature of drug-induced gingival overgrowth (Hall, 1997; Ilgenli et al., 1999; Kantarci et al., 1999).
Direct Effects of Phenytoin, Cyclosporin, and Nifedipine on Fibroblasts
Studies on the direct effects of phenytoin, nifedipine, and cyclosporin A on collagenous and non-collagenous extracellular matrix metabolism by gingival fibroblasts in culture have provided variable results (Schincaglia et al., 1992; Tipton et al., 1994; Newell and Irwin, 1997; James et al., 1998). The differences in these investigations depend in part on culture conditions (James et al., 1998, 2000). While cyclosporin A was found to increase glycosaminoglycan secretion by fibroblasts (Newell and Irwin, 1997), and nifedipine and phenytoin increased heparan levels (Saito et al., 1996), other studies failed to report any differences in vivo (Romanos et al., 1992, 1993; Rocha et al., 2000; Martins et al., 2003). Several studies demonstrate that these drugs inhibit gingival fibroblast extracellular matrix production and/or cell proliferation in vitro (Modéer et al., 1982, 1996; Salo et al., 1990; Stabellini et al., 1991; McKevitt and Irwin, 1995; Redlich et al., 1997). These findings are inconsistent with the in vivo characteristics of drug-induced gingival overgrowth. Taken together, these studies support the conclusion that direct regulation of extracellular matrix metabolism or proliferation of gingival fibroblasts by these drugs is probably not the primary mechanism responsible for gingival overgrowth. As noted below, deregulated cytokine balances may contribute more significantly to the development and maintenance of gingival overgrowth. Several studies more consistently show that cultured human gingival fibroblasts treated with phenytoin, cyclosporin A, and nifedipine increase production of fibroblast cytokines and prostaglandin E2 (Lerner et al., 1992; Modéer et al., 1992a,b, 2000; Brunius et al., 1993, 1996; Wondimu and Modéer, 1997), although lymphocytes and macrophages may be important sources of these factors in vivo (Modéer et al., 1989).
Cytokines and Drug-induced Gingival Overgrowth
Gingival tissues are generally in a state of injury and repair that involves repetitive cycles of production of chemotactic factors, inflammatory cell recruitment, and tissue resorption, replacement, and remodeling (Clark, 1998). Collagen turnover is unusually high in periodontal tissues (Sodek, 1978; Sodek and Ferrier, 1988). Wound healing and connective tissue turnover are largely controlled by chemokines and cytokines secreted by inflammatory cells such as macrophages and lymphocytes and, to a lesser degree, by fibroblasts. Proliferation and differentiation of connective tissue cells and production of extracellular matrix are controlled by cytokines that initiate signaling cascades mediated by specific receptors. In addition, extracellular matrix elements interact with cell-surface receptors, including integrins, that initiate or modulate signaling cascades (Clark, 1998; Arora et al., 2001). Recent studies demonstrate abnormally high levels of specific cytokines in gingival overgrowth tissues. These findings are of great interest, and suggest that substances that cause gingival overgrowth may do so by altering the normal balance of cytokines in gingival tissues. Cytokines and growth factors found at elevated levels in human drug-induced gingival overgrowth include interleukin-6 (IL-6), IL-1β, platelet-derived growth factor-B (PDGF-B), fibroblast growth factor-2 (FGF-2), transforming growth factor-β (TGF-β), and connective tissue growth factor (CTGF) (Williamson et al., 1994; Nares et al., 1996; Plemons et al., 1996; Saito et al., 1996; Dill and Iacopino, 1997; Iacopino et al., 1997; Atilla and Kutukculer, 1998; Sasaki and Maita, 1998; Hong et al., 1999; Myrillas et al., 1999; Buduneli et al., 2001; Uzel et al., 2001).
Origins of altered cytokine balances
There is a limited understanding of the mechanisms by which altered cytokine balances occur in drug-induced gingival overgrowth. A contributing factor may stem from immunomodulatory effects of the drugs. For example, the increased expression of the macrophage phenotype marker RM3/1 in phenytoin-induced gingival overgrowth is consistent with fibroproliferative disease (Iacopino et al., 1997). In contrast, macrophages in highly inflamed tissues express predominantly the 27E10 marker (Iacopino et al., 1997). Even though IL-1β levels have not been shown to be increased in cyclosporin-A-treated monocytes/macrophages, there was a significant up-regulation in PDGF-B in response to cyclosporin A and phenytoin (Plemons et al., 1996; Iacopino et al., 1997). Similarly, phenytoin, cyclosporin A, and nifedipine gingival overgrowth tissues contain subpopulations of macrophages and other inflammatory cells that differ from those in healthy control gingival tissues (Dahllof et al., 1985; Pernu et al., 1994; Cebeci et al., 1996, 1998; Pernu and Knuuttila, 2001; Bulut et al., 2002; Echelard et al., 2002). Studies on T- and B-lymphocytes showed that T-cells are increased in the peripheral blood of organ transplant patients with no apparent shift of subpopulations (Cebeci et al., 1998) and nifedipine increased lymphocyte counts in blood (Bullon et al., 2001), although these findings were not found in gingival tissues (Pernu and Knuuttila, 2001). A reduction in the number of Langerhans cells in nifedipine and cyclosporin A gingival overgrowth occurs and suggests a modification of an inflammatory reaction that influences the level of helper T-lymphocytes and cytokine profiles (Nurmenniemi et al., 2001). Inflammatory cell populations that are altered as a result of drug therapy are likely to modify the gingival tissue response. At this time, however, there is no consensus regarding functional relationships between and among drug therapies, the distribution of specific immune system cell subpopulations, and altered cytokine balances.
Functional studies
The occurrence of abnormal cytokine levels does not alone prove a functional relationship to gingival overgrowth. Studies have begun to investigate functional relationships between cytokines and gingival extracellular matrix metabolism. These studies seem likely to result in a greater understanding of the biological mechanisms that may be unique to human gingival tissues and may be relevant to the development of therapeutic strategies for either the prevention or treatment of gingival overgrowth. TGF-β1 is a cytokine secreted by many cell types, including macrophages, and it has an important regulatory function in collagen metabolism in connective tissues. TGF-β1 slowly stimulates collagen and lysyl oxidase biosynthesis in early-passage human gingival fibroblast cell cultures (Hong et al., 1999), whereas IL-1β, IL-6, and PDGF-BB have little or no effect, and FGF-2 is inhibitory (Hong and Trackman, 2001). In contrast, PDGF-BB and FGF-2 are potent mitogenic factors and contribute to the proliferation of gingival connective tissue and epithelial cells. The effects of TGF-β1 on gingival collagen and lysyl oxidase regulation are notable because the magnitude and kinetics of regulation are unexpectedly smaller and slower compared with studies on other connective tissue cells performed under the same conditions (Feres-Filho et al., 1995). To understand the unexpectedly slow kinetics of TGF-β1 on extracellular matrix synthesis in gingival fibroblasts, investigators in recent studies have focused on the presence and role of connective tissue growth factor (CTGF) as a possible matrix-stimulatory factor downstream of TGF-β1 in gingival overgrowth tissues. CTGF has been proposed to mediate the effects of TGF-β on extracellular matrix metabolism (Duncan et al., 1999). Before studies performed in gingival cells and tissues are summarized, information on CTGF and the emerging related CCN family of factors is first offered as background information.
CTGF and fibrosis
CTGF is found to occur at elevated levels in a variety of fibrotic pathologies, including the fibrous stroma of mammary tumors (Frazier and Grotendorst, 1997), chronic pancreatitis (di Mola et al., 1999), cataract formation (Wunderlich et al., 2000), nephropathy (Ito et al., 1998; Wang et al., 2001), systemic sclerosis (Sato et al., 2000), pulmonary fibrosis (Lasky et al., 1998; Sato et al., 2000), inflammatory bowel disease (Dammeier et al., 1998), bladder fibrosis due to outlet obstruction (Chaqour et al., 2002), brain fibrosis following injury (Hertel et al., 2000), atherosclerosis (Fan et al., 2000), and fibrotic skin disorders (Igarashi et al., 1996). CTGF alone does not promote fibrosis. This is illustrated by experiments in which the simultaneous application of CTGF and TGF-β1 is required for sustained skin fibrosis; neither factor alone was effective (Mori et al., 1999). CTGF is rapidly and potently induced by TGF-β1 in fibroblastic cells from a variety of different tissues, and contributes to the regulation of extracellular matrix genes (Blom et al., 2002).
CTGF is a member of the CCN family of factors (Oemar and Luscher, 1997; Brigstock, 1999; Moussad and Brigstock, 2000; Perbal, 2001; Blom et al.., 2002). The name of this family is derived from the first three family members identified: Cyr61, CTGF, and NOV. Additional members of this family include WISP-1 to -3. These factors have a highly conserved structure that consists of four domains containing 38 conserved cysteine residues. The four conserved domains (or modules) are related to other extracellular matrix proteins: Module 1 is similar to insulin-like growth factor (IGF) binding proteins; module 2 is similar to von Willebrand type C domain; module 3 is related to thrombospondin-1; and module 4 contains a putative cysteine knot that could promote dimerization of these factors. WISP-3 lacks the fourth module. The biological functions of this family of proteins include positive and negative regulation of proliferation and differentiation of connective tissue cells, and regulation of extracellular matrix accumulation. Structure/function studies are still in an early stage of development (Perbal, 2001; Blom et al., 2002). CTGF and cyr61 are closely related in structure, but promotion of extracellular matrix production or accumulation appears to be unique to CTGF (Chaqour et al., 2002). CTGF is expressed by endothelial cells, granulosa cells (Slee et al., 2001; Harlow et al., 2003), fibroblasts (Igarashi et al., 1996; Hong et al., 1999), mesangial cells (Goppelt-Struebe et al., 2001), chondrocytes (Igarashi et al., 1996), and osteoblasts (Xu et al., 2000), and occurs in biological fluids and tissues in low-molecular-weight forms that contain domains 3 and 4 that retain mitogenic activity (Brigstock et al., 1997). It seems possible that the pleiotropic nature of the CCN family of factors may be related to proteolytic processing events that unmask latent activities encoded by different functionally independent domains. Moreover, recent studies indicate that CTGF binds to other growth factors, resulting in either inhibition or stimulation of their activity. Thus, CTGF binds to vascular endothelial growth factor (VEGF) and bone morphogenetic protein-4 (BMP-4), resulting in inhibition of VEGF and BMP-4 activity, respectively; whereas CTGF binding to TGF-β1 is reported to be stimulatory (Abreu et al., 2002; Inoki et al., 2002). Matrix metalloproteinase (MMP) hydrolysis of CTGF/VEGF complexes results in release of active VEGF (Hashimoto et al., 2002). CTGF binds to αVβ3, α6β1, and other β3 integrins on different cell types and activates signaling cascades (Babic et al., 1999; Blom et al., 2002; Crean et al., 2002). Taken together, these studies support the idea that CTGF is a ‘matricellular’ factor that works in concert with growth factors, growth factor receptors, extracellular matrix, and extracellular matrix receptors (Bornstein, 2000). This view is consistent with the relatively weak direct stimulatory effects of CTGF on extracellular matrix production compared with the effects of pro-fibrogenic cytokines such as TGF-β1, and may account for the requirement for the simultaneous presence of both CTGF and TGF-β for sustained fibrosis (Mori et al., 1999). Integrin receptor-mediated signals in combination with growth factor receptor-mediated signals seem likely to work together to result in tissue fibrosis (Bornstein, 2000).
CTGF is developmentally regulated (Surveyor and Brigstock, 1999), and interesting studies regarding the presence and function of CTGF in developing tooth germs have been reported (Shimo et al., 2002). Inhibition of CTGF with a blocking antibody in murine tooth germ organ cultures inhibited tooth germ development and differentiation. Epithelial/mesenchymal interactions were found to be important for the expression of CTGF in tooth germs in tissue recombination experiments involving dental epithelium and mesenchyme, and TGF-β1 and BMP-2 were implicated as mesenchymal factors contributing to the maintenance of CTGF in tooth germs (Shimo et al., 2002).
CTGF in gingival overgrowth
As summarized above, CTGF contributes to fibrosis in many different tissues. The hypothesis was developed that CTGF could play a role in gingival overgrowth and fibrosis. Studies of CTGF regulation were initiated in vitro in human gingival fibroblast cultures, because increased collagen and lysyl oxidase biosynthesis induced by TGF-β1 occurred only at modest levels and with slow kinetics compared with effects seen in other cell types. The working hypothesis was that TGF-β1 might induce CTGF, which in turn would stimulate extracellular matrix production in gingival fibroblasts. Findings indicate that CTGF is rapidly and potently up-regulated by TGF-β1, and that CTGF stimulates insoluble collagen accumulation in human gingival fibroblast cultures (Hong et al., 1999). Moreover, a clinical study indicates that CTGF is present at elevated levels in phenytoin- and nifedipine-induced gingival overgrowth tissues, but not in cyclosporin-A-stimulated gingival overgrowth (Uzel et al., 2001). The finding of elevated CTGF in phenytoin-induced gingival overgrowth was obvious and clear, even after adjustment for the level of inflammation determined by histomorphometric analyses (Uzel et al., 2001). This study furthermore indicates that the more fibrotic tissues appear to contain the highest levels of CTGF. Analysis of the data obtained supports the notion that cyclosporin-A-induced gingival overgrowth tissues are significantly more inflamed and less fibrotic than phenytoin- or nifedipine-induced gingival overgrowth. Taken together, these findings identify clear and consistent molecular and cellular distinctions between and among phenytoin-, nifedipine-, and cyclosporin-A-induced gingival overgrowth tissues (summarized in the Table). It is apparent that gingival overgrowth is a clinical phenomenon that is heterogeneous with respect to the underlying biological mechanisms. Future studies will be necessary to identify additional molecular markers unique to specific forms of gingival overgrowth that will likely prove to be of functional significance in the etiology of different forms of this condition.
The mechanisms by which CTGF promotes fibrosis are being investigated by several different laboratories. There is no evidence for a specific unique dedicated CTGF signaling receptor in fibroblasts, and as noted, there is evidence for CTGF/integrin interaction with functional consequences (Babic et al., 1999; Blom et al., 2002; Crean et al., 2002). It is interesting that immunohistochemistry/immunocytochemistry studies suggest the presence of CTGF both in the extracellular matrix and within cells (Steffen et al., 1998; Surveyor et al., 1998; Hong et al., 1999; Surveyor and Brigstock, 1999; Kubota et al., 2000; Uzel et al., 2001). Thus, intracellular CTGF could potentially have function. A possible role in the inhibition of cell-cycle progression by intracellular CTGF has been suggested (Kubota et al., 2000). NOV, another CCN family member, likely has extracellular and intracellular functions. It binds to the extracellular matrix protein fibulin and is proposed to participate in extracellular matrix deposition and cell adhesion (Perbal et al., 1999); evidence for NOV in the nuclear envelope and a role for NOV as a co-factor in the transcriptional regulation of plasminogen activator inhibitor type 2 have been summarized (Perbal, 2001).
Unique Aspects of Gingival Fibroblast Metabolism
In some respects, it is surprising that CTGF is elevated in phenytoin-induced gingival overgrowth tissues. In human and rodent fibroblast cell lines, PGE2 is a potent and rapid down-regulator of CTGF, and this effect is mediated by elevated cAMP levels (Kothapalli et al., 1998; Duncan et al., 1999; Ricupero et al., 1999; Kothapalli and Grotendorst, 2000). Phenytoin stimulates prostaglandin E2 (PGE2) production by gingival fibroblasts (Modéer et al., 1992a), and it is well-known that human gingival tissues accumulate significant levels of prostaglandins including PGE2 (Arai et al., 1995). The fact that CTGF is elevated in phenytoin-induced gingival overgrowth tissues, therefore, is unexpected and raises the notion that human gingival tissues may be metabolically unique in their response to PGE2. The effects of PGE2 on fibroblasts are mediated principally by four receptors, EP1-EP4 (Coleman et al., 1994). PGE2 stimulation of cAMP levels is mediated primarily by the EP2 receptor (Sarrazin et al., 2001; Yu et al., 2002). Future studies will determine which PGE2 receptors are expressed by gingival fibroblasts, and whether phenytoin regulates either the function or the expression of prostaglandin receptors in gingiva in novel ways. It is very interesting to note that other recent studies support the notion that gingival fibroblast extracellular matrix metabolism has unique aspects. In healing adult human gingiva, MMP-13, and not MMP-1, is highly expressed by gingival fibroblasts. In contrast, MMP-1 is predominantly expressed in adult dermal wounds. Moreover, MMP-13 is strongly up-regulated by TGF-β1 in human gingival fibroblasts, but is down-regulated in human dermal fibroblasts grown under the same conditions (Ravanti et al., 1999; Leivonen et al., 2002). MMP-13 is a collagenase with wide substrate specificity, whereas MMP-1 is a collagenase more restricted to hydrolyzing fibrillar collagens. Differential regulation of these two MMP genes is likely to be of biological importance, and may be related to the generally lower tendency for scar formation in oral tissues (Ravanti et al., 1999). In addition, integrin expression patterns differ between gingival fibroblasts and dermal fibroblasts, whereas periodontal ligament fibroblasts and gingival fibroblasts are more similar to each other in this respect (Palaiologou et al., 2001). It is interesting to note that human gingival fibroblasts and periodontal ligament fibroblasts express a variety of different genes differentially, including members of the CCN family of genes (Han and Amar, 2002). Unique aspects of gingival fibroblast extracellular matrix metabolism seem likely to contribute to the etiology of gingival overgrowth. In addition, metabolic uniqueness of gingival cells may help explain the striking tissue specificity of drug-induced gingival overgrowth.
Cyclosporin-A-induced gingival overgrowth is an interesting example of tissue-specific mechanisms that are not fully understood at this time. A major and serious side-effect of cyclosporin A therapy is kidney fibrosis, which can result in kidney failure (Myers et al., 1984). Cyclosporin A stimulates levels of circulating TGF-β in vivo (Khanna et al., 1997), and enhances TGF-β production by renal cells and lymphocytes (Ahuja et al., 1995; Prashar et al., 1995; Young et al., 1995; Wolf et al., 1996). This results in increased collagenous extracellular matrix synthesis and deposition in the glomeruli of the kidney, as demonstrated by studies with anti-TGF-β1 antibodies that block renal fibrosis and renal dysfunction (Shihab et al., 1996; Islam et al., 2001). Taken together, these studies suggest that cyclosporin A stimulates TGF-β production that, in turn, leads to kidney fibrosis and nephropathy. Based on these findings, it seemed reasonable to expect that TGF-β1 and its downstream target CTGF would be expressed at high levels in cyclosporin-induced gingival overgrowth (Wondimu et al., 1997). Contrary to these expectations, cyclosporin-induced gingival overgrowth tissues are highly inflamed (Echelard et al., 2002), do not express high levels of TGF-β or CTGF, and are not the most fibrotic tissues (Uzel et al., 2001). These findings are surprising and indicate that oral bacteria and gingival cells and tissues must interact in unique ways in subjects receiving cyclosporin A that results in relatively greater inflammation and cellularity compared with other forms of gingival overgrowth. The biological mechanisms responsible for this phenomenon must be unique to gingival tissues and cells and are currently under investigation.
Gingival Overgrowth and Diminished Tissue Resorption
Diminished tissue resorption as a mechanism contributing to drug-induced gingival overgrowth has received some attention (Thomason et al., 1998). Connective tissue turnover in gingival tissues is high, and destruction of the extracellular matrix occurs as a result of elaboration of extracellular proteinases, reduced MMP activity, and by phagocytosis and intracellular destruction of extracellular matrix components by lysozomal enzymes (McCulloch and Knowles, 1993; Thomason et al., 1998). In vitro studies have shown that phenytoin and cyclosporin A inhibit production of the lysozomal proteinase cathepsin L, but not cathepsin B, by human gingival fibroblasts (Yamada et al., 2000). Cyclosporin may also inhibit phagocytosis of type I collagen in vitro and in vivo (Kataoka et al., 2000; Arora et al., 2001). Consistent results were found in cathepsin-L-deficient mice that exhibited enlarged gingival epithelial and connective tissues (Nishimura et al., 2002). The authors note that humans lacking lysozomal enzymes as a result of the rare genetic disease I-cell disease or mucolipidosis also have gingival overgrowth (Nishimura et al., 2002). Thus, inhibition of phagocytosis or of lysozomal enzymes appears likely to be a mechanism that could contribute to gingival overgrowth. The possible role of inhibited phagocytosis and lysozomal enzyme activity in contributing to gingival overgrowth is interesting and requires further study.
Inherited Gingival Overgrowth
The occurrence of inherited forms of gingival overgrowth provides two important experimental strategies to aid in our understanding of the mechanisms of gingival overgrowth that may also provide insights into the mechanisms of drug-induced gingival overgrowth. First, there is the opportunity to study the phenotype of connective tissue cells in vitro from affected individuals without the complications of direct or indirect and transient effects of drugs. Second is the opportunity to perform genetic linkage analysis in affected families with the ultimate goal of identifying mutated genes that contribute to gingival overgrowth. Both experimental strategies have been pursued. Studies have found that gingival fibroblasts cultured from affected individuals generally produce higher levels of TGF-β1, have higher rates of proliferation, and respond to TGF-β1 by making increased levels of extracellular matrix (Tipton et al., 1997; Tipton and Dabbous, 1998). Thus, gingival fibroblasts from affected individuals have an autocrine pathway involving high production of TGF-β coupled to a robust response to this factor, resulting in increased production of connective tissue cells and extracellular matrix.
Genetic linkage studies of large families have identified more than one locus related to gingival overgrowth in hereditary gingival fibromatosis (Hart et al., 1998, 2000; Shashi et al., 1999). These findings support the notion that not all forms of gingival overgrowth are the same, and that more than one biological mechanism is likely to result in gingival overgrowth. A detailed study of a large Brazilian family has identified a specific gene mutation that segregates with the hereditary gingival fibromatosis phenotype (Hart et al., 2002). This study is the first to link a specific gene mutation to a phenotype of gingival overgrowth. The Sos1 protein is a GTP exchange factor required for the activity of ras proteins. Thus, Sos1 facilitates the exchange of GDP for GTP on the active site of ras, and thereby activates ras proteins. Ras proteins are involved in signal transduction pathways initiated by tyrosine kinase receptors in all cell types (Shapiro, 2002). The signaling pathway (Fig. 1) is typically initiated by the binding of a ligand to a cell-surface tyrosine kinase receptor, resulting in phosphorylation of specific tyrosine residues in the cytoplasmic domain of the receptor. Intracellular adapter proteins, most notably Shc and Grb2, then bind and recruit Sos1 to the membrane receptor. Ras is bound to the inner surface of the plasma membrane and contains either GDP or GTP, and recruited Sos1 stimulates the exchange of GTP for GDP on ras, thereby activating ras. Ras bound to GTP stimulates the activity of downstream protein kinases and other small GTP-binding proteins, whereas ras is inactive if it is bound to GDP. The downstream protein kinases/GTP proteins include the MAP kinase family, phoshatidylinositol-3 kinase, and Rho-proteins. These activities ultimately control the activity of transcription factors and co-activators that regulate the expression of a variety of genes required for proliferation and differentiation in different cell types (Shapiro, 2002). Somatic mutations of ras that are constitutively active are oncogenic and contribute to cell transformation and cancer in a variety of tissues (Bos, 1989). The Sos1 mutation linked to gingival overgrowth is a single nucleotide insertion that causes a frame shift and premature termination. The resulting predicted mutant Sos1 protein would contain an abnormal and truncated C-terminus. This mutant protein product is proposed to be constitutively active (Hart et al., 2002). This prediction suggests that increased ras activity would occur in individuals who carry this mutation and would in some way lead to gingival overgrowth. Because this is an inherited mutation and not a somatic mutation, all cells in affected individuals contain mutated Sos1. Therefore, at present, we do not know the mechanism by which this mutation contributes to gingival overgrowth in particular. For example, it is unknown why this mutation does not result in obvious abnormalities in other tissues in affected individuals. A general hypothesis is that, as noted earlier in this review, metabolic pathways in gingival cells and tissues appear to be unique in certain respects. Thus, the Sos1 mutation may affect gingival connective tissue cell biology in unique ways. Studies of the biological activity of the mutated Sos1 protein in gingival cells and in cells from other human tissues could, therefore, be potentially very informative.
Fibroblast Subpopulations and Fibroblast Differentiation
Fibroblast heterogeneity and variations in the distribution of fibroblasts with different phenotypes have been suggested to contribute to drug-induced and inherited forms of gingival overgrowth (Hassell, 1981; Hassell and Stanek, 1983; Pagliarini et al., 1995). The presence of myofibroblasts in gingival overgrowth tissues has been reported (Dill and Iacopino, 1997), and these are highly differentiated cells that have a strong synthetic phenotype (Powell et al., 1999a,b). The presence and role of myofibroblasts in systemic sclerosis and skin fibrosis is known (Jelaska et al., 1996, 1999). A final outcome of tissue overgrowth and fibrosis is characterized by the presence of fibroblasts with an activated synthetic phenotype, and the mechanisms responsible for the presence of these cells may involve reciprocal interactions between lymphocytes and fibroblasts (Fries et al., 1994). Mechanical forces contribute in some way to fibroblast selection and differentiation (Kessler et al., 2001; Schild and Trueb, 2002). As discussed above, deregulated cytokine expression and unique aspects of gingival fibroblast metabolism are all likely to be important contributing factors in gingival overgrowth and development of highly synthetic or proliferative fibroblast phenotypes (Schmitt-Graff et al., 1994; Vaughan et al., 2000). In addition, a reduced rate of apoptosis is reported to contribute to the accumulation of gingival fibroblasts (Fujimori et al., 2001), perhaps with a greater synthetic or proliferative phenotype in nifedipine-induced gingival overgrowth (Kessler et al., 2001; Schild and Trueb, 2002), though this is a novel and understudied area at this time.
Summary of Current Understanding
A scheme summarizing some functional relationships in gingival overgrowth is presented in Fig. 2. Recent studies summarized indicate that molecular markers and clinical features of gingival overgrowth differ depending on the responsible medication. Similarly, multiple different genetic loci have been liked to inherited forms of gingival overgrowth, supporting the notion that the biological origins for gingival overgrowth are complex. Data from several different laboratories have provided evidence that cytokine and growth factor balances are altered in gingival overgrowth tissues, including CTGF, a member of the interesting CCN family of factors. Cytokine-dependent alterations in extracellular matrix metabolism appear to be of functional importance to gingival overgrowth. Abnormal differentiation of cells, resulting in accumulation of fibroblasts with a pathologic range of proliferative and synthetic phenotypes, could result from deregulated cytokines. New data, in addition, support that aspects of gingival fibroblast extracellular matrix metabolism are unique, and may help to explain the tissue specificity of gingival overgrowth. Further studies are required to define unique metabolic aspects of gingival extracellular matrix metabolism; and a greater understanding of interactions between and among medications, the innate and acquired immune response, cytokines and growth factors, and gingival epithelial and connective tissue cells will provide more detailed molecular and mechanistic information that will ultimately have therapeutic relevance to the prevention and treatment of gingival overgrowth. The accomplishment of these objectives will contribute to the improvement of oral and systemic health.
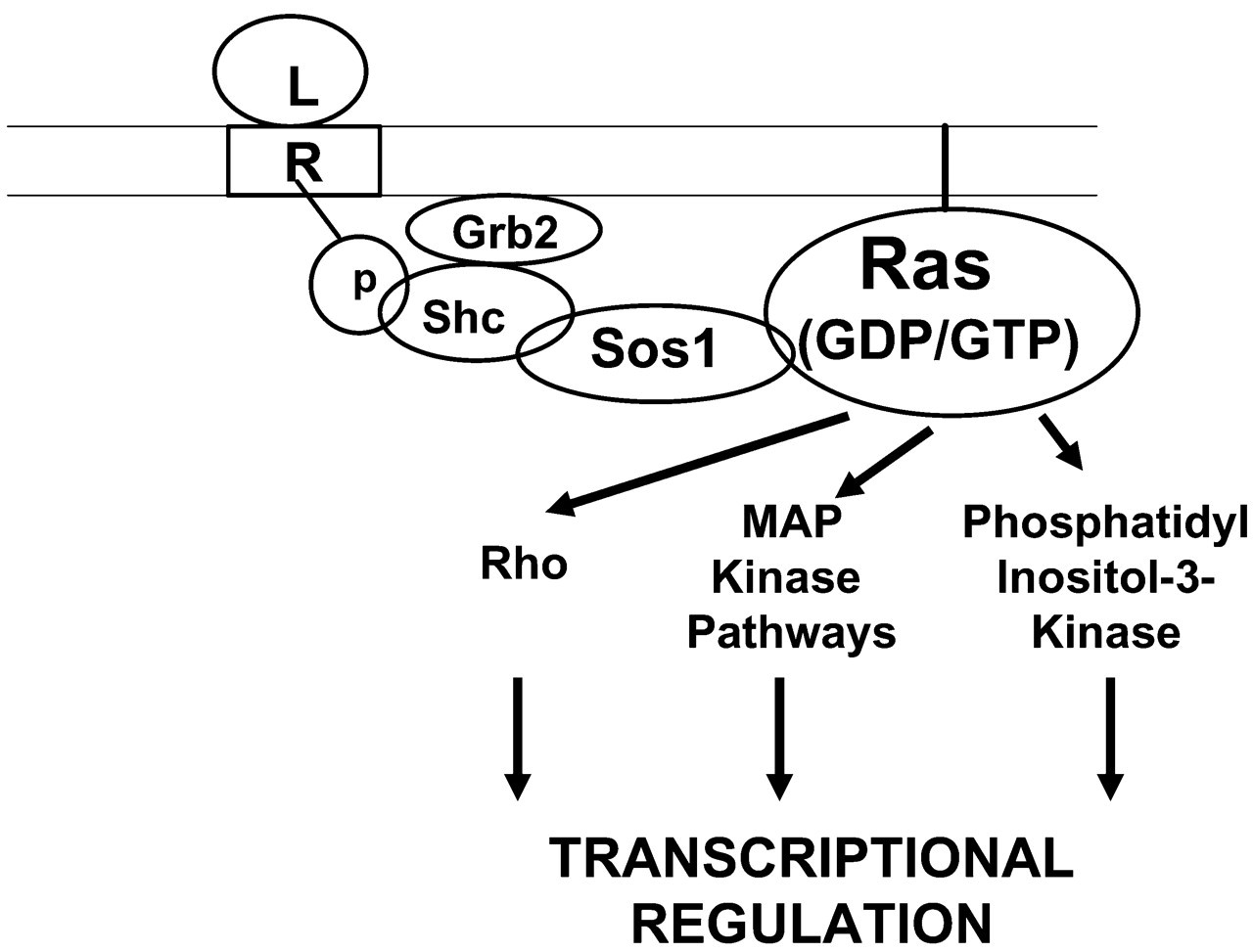
Elements of tyrosine kinase receptor signal transduction pathways showing the role of Sos1. Ligand (L) binds to tyrosine kinase receptor (R), leading to phosphorylations in the cytoplasmic domain of the receptor (p). This recruits adapter proteins (Grb2 and Shc) and Sos1. Bound Sos1 then catalyzes the substitution of GTP for GDP on plasma-membrane-bound Ras proteins. Active GTP/Ras then activates protein kinases, ultimately resulting in activation and translocation of transcription factors to the nucleus, and regulation of transcription of specific target genes.
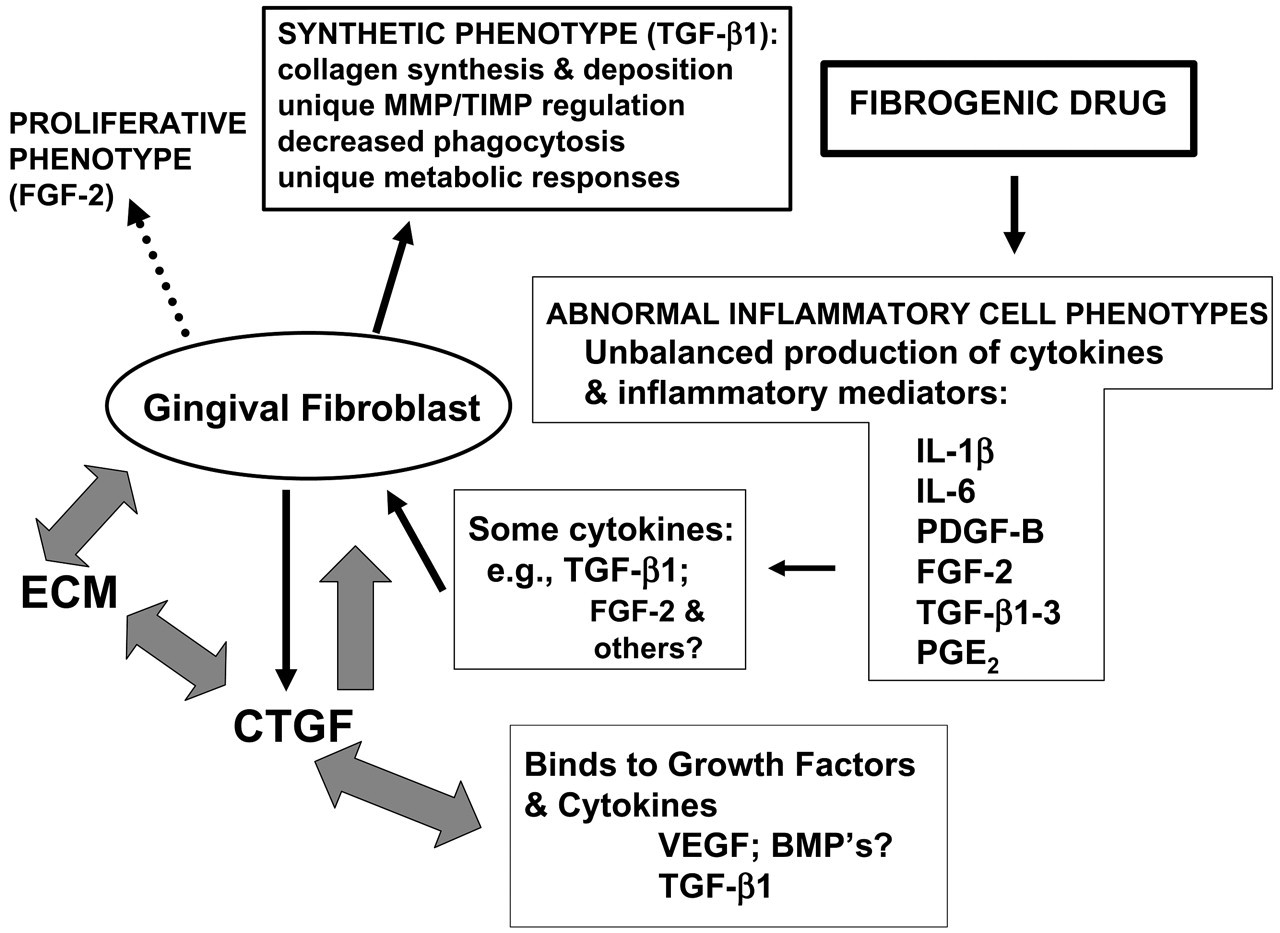
Summary of functional interactions in drug-induced gingival overgrowth. Fibrogenic drugs (phenytoin, nifedipine, cyclosporin A) are envisioned to alter the phenotypes of inflammatory cells (principally lymphocytes and macrophages), resulting in the observed abnormal balances of cytokines and inflammatory mediators. This would result in distortion of normal responses to bacterial insults and injury, as follows: A subset of cytokines would influence gingival fibroblast extracellular matrix metabolism and proliferation. For example, in the case of TGF-β1 overexpression in phenytoin-induced gingival overgrowth, fibroblasts produce and accumulate CTGF that has direct and indirect effects on extracellular matrix production, cell/extracellular matrix (ECM) interactions, and activity of other growth factors and cytokines, and gingival fibroblast extracellular matrix production (block arrows). By contrast, increased FGF-2 is seen as principally a mitogenic factor that stimulates gingival fibroblast proliferation. In addition, unique aspects of gingival fibroblast metabolism are believed to confer tissue-specific features of extracellular matrix metabolism in gingiva.
Footnotes
Acknowledgements
This work was supported by NIH/NIDCR grant DE 11004 to PCT. The authors are grateful to Dr. Mehmet Ilhan Uzel for suggestions and critical reading of this manuscript.