
Abstract
Osteoclasts are tissue-specific polykaryon bone-resorbing cells derived from the monocyte/macrophage hematopoietic lineage with specialized functions required for the adhesion of the cells to bone and the subsequent polarization of the cell membrane, secretion of acid to dissolve mineral crystals, and release of proteolytic enzymes to degrade the extracellular matrix proteins. Most pathological conditions in the skeleton lead to loss of bone due to excess osteoclastic bone resorption, including periodontal disease, rheumatoid arthritis, and osteoporosis. In rare cases, most of them genetic, patients with osteopetrosis exhibit sclerotic bone due either to a lack of osteoclasts or to non-functional osteoclasts. Mainly because of phenotypic findings in genetically manipulated mice or due to spontaneous mutations in humans, mice, and rats, several genes have been discovered as being crucial for osteoclast formation and activation. Recent breakthroughs in our understanding of osteoclast biology have revealed the critical roles in osteoclast differentiation played by RANKL, RANK, and OPG, three novel members of the tumor necrosis factor ligand and receptor superfamilies. The further study of these molecules and downstream signaling events are likely to provide a molecular basis for the development of new drugs for the treatment of diseases with excess or deficient osteoclastic bone resorption.
(1) Introduction
Metabolism of bone tissue in the skeleton, including jaw bones, is dependent on the concerted actions of the bone formation and bone resorption processes. During embryonic development and post-natal growth, the amount of bone formed exceeds that being resorbed. In the adult skeleton, which is continuously remodeled, the two processes are tightly coupled and in balance, resulting in the preservation of bone mass obtained during the growth period. In pathological situations, bone formation and bone resorption are often uncoupled, resulting in either loss of bone—as in periodontal bone disease, osteoporosis, rheumatoid arthritis, and most malignant tumors—or in gain of bone—as in osteopetrosis, certain malignant tumors, and some inflammatory conditions.
The physiological remodeling of bone takes place in so-called ‘bone multi-cellular units’ (BMUs) and is initiated by recruitment, formation, and activation of bone-resorbing osteoclasts. These cells resorb a given volume of bone, and, subsequently, the Howship’s resorption lacunae are filled with new bone formed by recruited and activated osteoblasts. Such BMUs are present both at the surfaces of trabecular and cortical bone and in the Haversian canals of cortical bone but are more frequent in trabecular bone. This is the reason a metabolic bone disease such as osteoporosis primarily affects bones with a large proportion of trabecular bone. It has been estimated that, under normal conditions, 1 or 2 × 106 BMUs are present in the adult skeleton.
Since remodeling of bone in pathological conditions mostly results in increased bone resorption, much of the interest in bone cell biology has been focused on osteoclasts. Many different systemic hormones and local growth factors/cytokines have been demonstrated to stimulate or inhibit osteoclast formation and/or function. Also, recently, to this list of osteotropic factors have been added the signaling molecules present in the peripheral nervous system and mechanisms controlled by the central nervous system (Baldock et al., 2002; Lerner and Lundberg, 2002. Takeda et al., 2002).
Already during the late 1970s, it was shown that osteoclasts are derived from haematopoietic tissues and are in fact leukocytes. We now know that osteoclasts are formed by fusion of mononuclear progenitors of the monocyte-macrophage lineage. During the last decade, it has become evident that osteoblasts in the periosteum, and osteoblast-like stromal cells in hematopoietic tissues, control osteoclast formation/activation via cell-to-cell contacts with the progenitor cells. During past years, the molecules responsible for the interaction between these cells have been found to be members of the tumor necrosis factor (TNF) ligand and receptor superfamilies. Thus, the expression of receptor activator of nuclear factor-κB ligand (RANKL) on the surfaces of stromal cells/osteoblasts and the activation of its cognate receptor RANK on the surfaces of hematopoietic cells are required for osteoclast formation and activation (Fig. 1). This interaction can be blocked by the soluble ‘decoy’ receptor osteoprotegerin (OPG) secreted by stromal cells/osteoblasts. Mainly due to expected, and sometimes unexpected, phenotypic findings in mice with specific targeted gene deletions, we have also acquired information on the critical importance for the differentiation and function of osteoclasts of other molecules expressed in stromal cells/osteoblasts and osteoclast progenitor cells. It is the aim of the present review to summarize, briefly, the knowledge about the molecules involved in osteoclast differentiation and activation and to focus, in more detail, on the TNF ligand and receptor-like molecules.
(2) Osteoclast Differentiation
Mononuclear osteoclast progenitor cells are derived from stem cells present in hematopoietic tissues. These cells proliferate and differentiate to a mononuclear progenitor cell that enters the circulation and then, by unknown homing mechanisms, eventually enters the periosteum and endosteum covering all bone surfaces. Subsequently, the precursor cells can undergo further differentiation and finally, at the bone surface, fuse to the multinucleated osteoclast. Whether or not osteoclasts present on bone surfaces within bone marrow really reach the bone via circulation, or if the osteoclast progenitor cells simply migrate in the bone marrow cavity to the bone surfaces, is not fully understood. The local environment of bone can be mimicked ex vivo by the culturing of bone marrow, or by the co-culturing of stromal cells/osteoblasts with either spleen cells or mononuclear leukocytes from peripheral blood. In such systems, the cultured osteoclast precursor cells can be induced to differentiate and fuse to functionally active multinucleated osteoclasts after stimulation with, e.g., parathyroid hormone (PTH) or 1,25(OH)2-vitamin D3 (D3). Gene deletion experiments in mice have shown that commitment of osteoclast progenitor cells to functionally active mature osteoclasts requires activation of transcription factors, hematopoietic cytokines, cell-surface receptors, proteolytic enzymes, phosphatases, receptor-associated molecules, and kinases. Some of these molecules are expressed in stromal cells/osteoblasts and others in the osteoclast precursor cells (Fig. 1).
Mice deficient in the transcription factor PU.1 lack both macrophages and osteoclasts, because of the absence of common early precursor cells for macrophages and osteoclasts (Tondravi et al., 1997). Consequently, the osteopetrotic phenotype of PU.1 −/− mice can be rescued by bone marrow transplantation.
Activation of the c-fms receptor by its cognate ligand macrophage colony-stimulating factor (M-CSF) is necessary for the proliferation and survival of macrophages/osteoclast progenitor cells, and loss of function mutation in the M-CSF gene in op/op mice is the reason for the lack of osteoclasts in these mice and their osteopetrotic phenotype (Yoshida et al., 1990). Since M-CSF is expressed by stromal cells/osteoblasts, op/op mice are not cured by bone marrow transplantation. The signaling pathway of M-CSF includes mitogen-activated protein kinases (MAPK)-induced phosphorylation of Mitf and TFE3, two closely related helix-loop-helix transcription factors of which Mitf has been linked to osteopetrosis in mi/mi mice (Weilbaecher et al., 2001). Interestingly, the osteopetrotic phenotype of the op/op mice is lost over time due to enhanced expression of granulocyte-macrophage colony-stimulating factor (GM-CSF), indicating a redundancy of the M-CSF and GM-CSF genes.
The transcription factor AP-1 is a heterodimeric protein consisting of Fos proteins (c-Fos, FosB, Fra-1, and Fra-2) and Jun proteins (c-Jun, JunB, and JunD). Unexpectedly, mice deleted of the c-Fos gene exhibit an osteopetrotic phenotype due to lack of osteoclast progenitor cells (Wang et al., 1992; Grigoriadis et al., 1994). The osteopetrotic phenotype of c-Fos-deficient mice can be cured by bone marrow transplantation. The arrest of osteoclast formation in the c-Fos −/− mice is associated with increased numbers of macrophages, indicating that c-Fos acts downstream of PU.1. The lack of osteoclastogenesis in spleen cell cultures from c-Fos −/− mice can be rescued by in vitro transfection by all forms of Fos proteins, but not by Jun transfection, with Fra-1 having the highest activity (Matsuo et al., 2000). In line with these observations, transgenic mice expressing Fra-1 prevent the osteopetrotic phenotype of c-Fos-deficient mice. RANKL induces c-Fos-dependent transcription of Fra-1, indicating that Fra-1 is acting distal to c-Fos, which is also shown by the fact that Fra-1 expression is blunted in c-Fos −/− mice. Interestingly, RANKL-induced expression of interferon-β (IFN-β) in stromal cells/osteoblasts acts in a negative feedback manner and inhibits osteoclast formation by decreasing RANKL-stimulated c-Fos expression (Takayanagi et al., 2002a). The IFN-β-induced inhibition of osteoclastogenesis can be rescued by suppressors of cytokine signaling (SOCS)-1 and-3, which also are up-regulated by RANKL (Hayashi et al., 2002). Mice which lack responsiveness to IFN-β develop osteoporosis (Takayanagi et al., 2002a).
As described in detail below, deletions of the RANKL or RANK genes result in the absence of osteoclasts due to arrested differentiation of M-CSF expanded osteoclast progenitor cells. Deletion of the OPG gene, resulting in loss of the RANKL-inhibitory OPG, gives rise to mice with early-onset osteoporosis. The differentiation of osteoclast progenitor cells is also blocked by deletion of the gene for TNF receptor-associated factor 6 (TRAF6) (Naito et al., 1999), and by double knockout of the NF-κB proteins p50 and p52 (Franzoso et al., 1997; Iotsova et al., 1997), which is not unexpected, since TRAF6 and NF-κB are part of the RANK signaling pathway. The osteopetrotic phenotype of p50/p52 −/− and TRAF6 −/− mice can be overcome by bone marrow transplantation, indicating the importance of these molecules in the osteoclast progenitor cells. Recently, Battaglino et al. (2002), in a cDNA microarray screening project, found the proto-oncogene c-Myc to be a downstream target in RANK signaling in RAW 264.7 cells, and that expression of a dominant-negative Myc in these cells blocked RANKL-induced osteoclast formation.
The differentiation of mononuclear osteoclast progenitor cells to mature osteoclasts involves fusion to multinuclear cells and their polarization of the cell membrane adjacent to bone, required for development of the sealing zone and the ruffled border (Fig. 2). Attachment of osteoclasts to bone extracellular matrix in the sealing zone is associated with the presence of F-actin rings and has been suggested to be mediated by the integrin αvβ3, preferentially expressed in the sealing zone area, and the RGD sequence in osteopontin and bone sialoprotein. The importance of the αvβ3 expression is shown by the observations that RGD-containing peptides can block bone resorption in vitro, and that, although β 3−/− mice can form multinucleated osteoclasts, the osteoclasts are less polarized, as assessed by a lack of actin rings and disorganization of the ruffled border (McHugh et al., 2000). The functional importance of these abnormalities is demonstrated by the facts that blood calcium is decreased, bone mass increased, and osteoclasts in β 3 −/− mice are less effective in resorbing bone on dentin slices in vitro. Interestingly, it has been suggested that ligand binding to αvβ3 may not only be part of the attachment of osteoclasts to extracellular matrix, but also may be involved in intracellular signaling. Thus, αvβ3 activation is associated with changes of intracellular calcium and activation of c-src- and c-src-dependent phosphorylation of proline-rich tyrosine kinase-2 (Pyk-2). The latter is a kinase related to the focal adhesion kinase (FAK), which is suggested to be involved in formation of the sealing zone, as well as to the activation of phosphatidylinositol 3-hydroxyl kinase (PI3-kinase) (Hruska et al., 1995; Chellaiah et al., 1998; Duong et al., 1998; Zhang et al., 2002; Wang et al., 2003). The view that αvβ3 is not only of importance for anchoring osteoclasts to the extracellular matrix is supported by the observation that transfection of the full-length β3-integrin gene rescues the impaired osteoclast function in β 3 −/− mice, whereas transfection with a truncated form of β3, lacking the cytoplasmic domain, does not have this activity (Feng et al., 2001). Interestingly, transfection with a construct containing the S752P substitution in the β3 cytoplasmatic domain, characteristic of the bleeding disorder Glanzmann’s thrombasthenia, fails to restore the function of β 3 −/− osteoclasts. The crucial role of ruffled border formation in osteoclast activity is illustrated by the finding that c-src-deficient mice have normal numbers of multinucleated osteoclasts, but the cells lack the ruffled border, and, as a consequence, the osteoclasts fail to resorb bone, and the c-src −/− mice exhibit an osteopetrotic phenotype (Soriano et al., 1991; Boyce et al., 1992). These mice can be cured by bone marrow transplantation.
The hypothesis that interaction between α v β 3 and osteopontin is important for bone resorption has also been examined in osteopontin-deficient mice. These mice exhibit no skeletal phenotype under normal conditions. In favor of the hypothesis, however, it has been found that osteopontin −/− mice are resistant to ovariectomy-induced bone loss (Yoshitake et al., 1999). Furthermore, bone resorption and osteoclast formation induced by PTH in cultured mouse calvariae are absent in calvariae from osteopontin knockout mice (Ihara et al., 2001). Thus, it is evident that osteopontin has a role in not only osteoclast polarization, but also in osteoclast recruitment. The molecular mechanism involved in this latter process is unknown.
The bone-resorbing activity of a fully differentiated multinucleated osteoclast that is attached to bone and has developed a ruffled border involves mechanisms for dissolution of hydroxyapatite crystals and enzymatic degradation of bone matrix proteins. The process is initiated by secretion of protons mediated by a vacuolar type of H+-ATPase in the ruffled border. By this proton pump, a pH of ~ 4.5 is created in the microenvironment present in the Howship’s resorption lacunae, and bone mineral will be dissolved. The osteoclastic proton pump seems to contain a specific subunit, termed OC-116kD, which has been cloned. Targeted disruption of its gene (Atp6i) results in mice which can form osteoclasts, but the cells lose their capacity to generate extracellular acidification (Li et al., 1999). These mice exhibit a severe form of osteopetrosis. Atp6i −/− mice have normal intracellular lysosomal proton pump activity in osteoclasts and no disturbances in the liver lysosomes, or proton pump transport in kidney microsomes. Deletion in this gene has been found in the osteosclerotic mutant oc/oc mice (Scimeca et al., 2000). Several mutations in the human Atp6i gene, the vast majority of which result in loss of function, have recently been found in patients with autosomal-recessive osteopetrosis (Sobacchi et al., 2001; Michigami et al., 2002; Scimeca et al., 2003; Taranta et al., 2003).
To provide electroneutrality of acid secretion, chloride channels are present together with the H+-ATPase in osteoclastic ruffled-border membranes. Recently, the critical importance of the function of osteoclasts of one of the 9 known mammalian chloride channel genes was shown. Deletion of the gene for the ubiquitously expressed chloride channel ClC-7 resulted in mice with severe osteopetrosis and retinal degeneration (Kornak et al., 2001). Osteoclasts from ClC-7 −/− mice differentiate normally and attach to bone surfaces, but are unable to create resorption lacunae because of a failure to secrete acid, although the H+-ATPase is expressed normally. Mutations in the human ClC-7 gene cause osteopetrosis in patients (Cleiren et al., 2001; Kornak et al., 2001; Campos-Xavier et al., 2003).
The demineralized bone in the resorption lacunae is degraded by proteolytic enzymes. The knowledge of which enzymes are involved in degradation of the different proteins in bone extracellular matrix is very limited. It is well-recognized, however, that cysteine proteinases play a crucial role, and, in general, cysteine proteinase inhibitors are potent inhibitors of bone resorption in vitro and in vivo. Cathepsin K is a cysteine proteinase with collagenolytic activity abundantly expressed in osteoclasts. Deletion of the cathepsin K gene results in mice with an osteopetrotic phenotype (Saftig et al., 1998; Gowen et al., 1999). In the resorption area beneath the osteoclasts, demineralized, but undegraded, bone matrix can be seen, indicating that the defect is primarily due to a lack of extracellular collagenolytic activity. This feature is also characteristic of resorption lacunae in the human osteopetrotic disease pycnodysostosis, which is linked to several mutations in the cathepsin K gene (Gelb et al., 1996). The possible role of matrix metalloproteinases (MMP) in osteoclastic resorption is less clear. An interesting hypothesis put forward by Delaisse and co-workers suggests that MMPs are required for the invasion of osteoclasts through the extracellular matrix, rather than for their activity in the Howship’s lacunae (Ensig et al., 2000). It has also been suggested that the role of MMPs in bone resorption is to be involved in ‘cleaning’, performed by the lining cells, of collagen fibrils left in the resorption lacunae by osteoclasts (Everts et al., 2002).
Recently, we have found that cysteine proteinases are involved not only in the bone-resorbing activity of osteoclasts, but also in osteoclast differentiation. Thus, cystatin C and a peptidyl derivative synthesized to mimic part of the proteinase-binding site of cystatin C (Z-RLVG-CHN2) inhibit osteoclast formation in mouse bone marrow cultures stimulated by PTH, D3, or IL-6 and in spleen cell cultures stimulated by M-CSF and RANKL. The effect is exerted at a late stage of the osteoclast progenitor cell differentiation. The enzyme(s) and mechanisms involved is/are presently unknown (Brage et al., 2004).
(3) Importance of Osteoblast/Stromal Cells in Osteoclast Differentiation
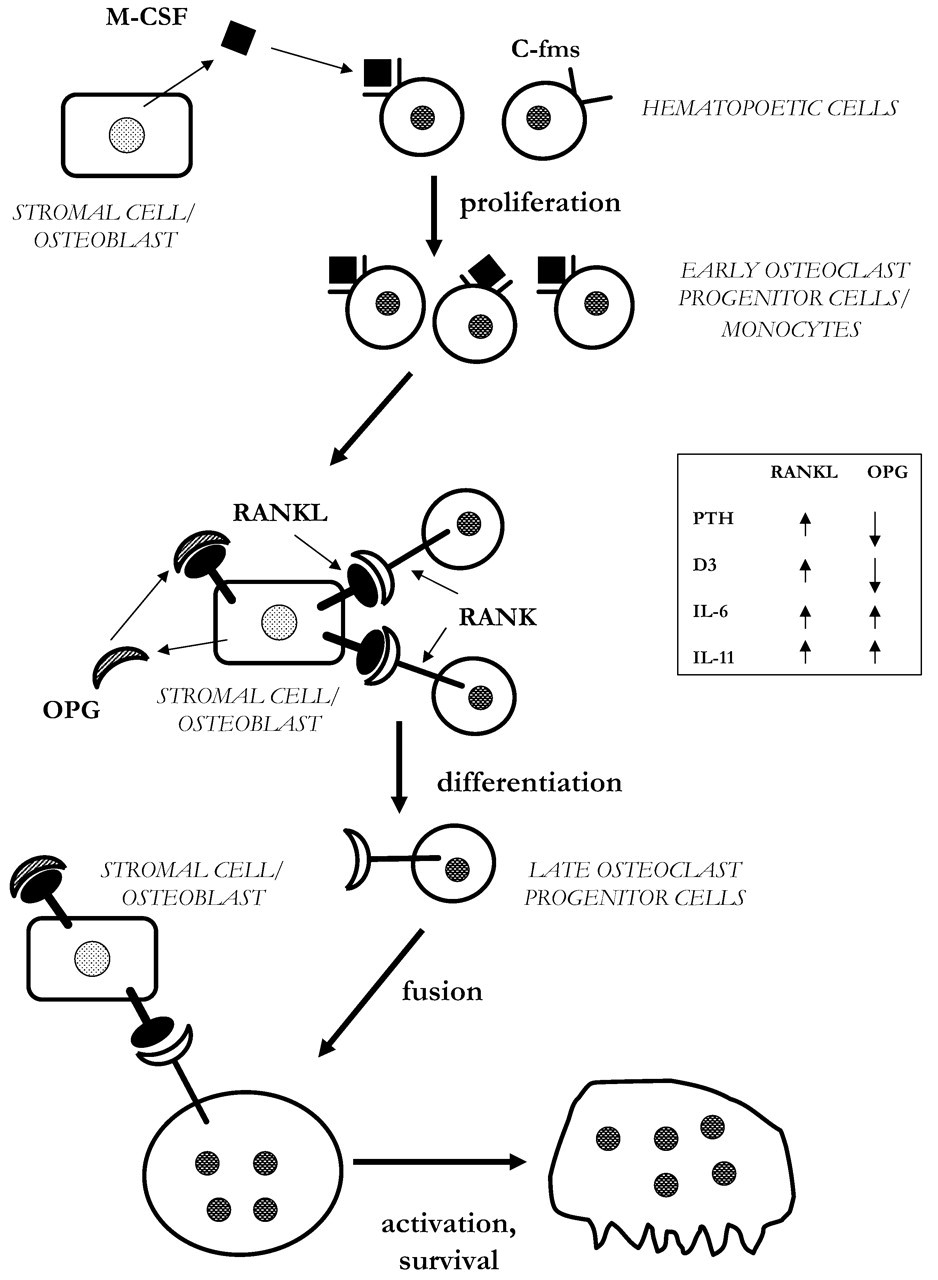
The crucial role of stromal cells/osteoblasts in osteoclast development was initially indicated by the observation that receptors for the bone-resorbing hormones PTH and D3 were present not in osteoclasts or their precursor cells, but in osteoblasts. Primarily by the use of co-culture systems with stromal cells/osteoblasts and osteoclast precursors, it has become clear that stromal cells/osteoblasts play a pivotal role in supporting osteoclastogenesis and that cell-to-cell contact between the two cell types is required (Fig. 2). Gene deletion experiments have very elegantly demonstrated the indispensable role of osteoblasts, not only for osteoclast formation stimulated by PTH and D3, but also for the osteoclastogenic effects of interleukin-6 (IL-6) and prostaglandin E2 (PGE2). Thus, when osteoblasts isolated from PTH receptor (PTHR1) knockout mice, vitamin D3 receptor (VDR) knockout mice, or mice lacking the EP4 receptor for PGE2 were co-cultured with spleen cells from wild-type mice, no osteoclasts were formed in response to the ligand for these receptors (Liu et al., 1998; Takeda et al., 1999; Sakuma et al., 2000). In contrast, when spleen cells from PTHR1 −/− mice, VDR −/− mice, or EP4 −/− mice were co-cultured with osteoblasts from wild-type mice, osteoclasts were formed in response to PTH, D3, or PGE2. These observations demonstrate that it is the expression of the receptors for PTH, D3, and PGE2 in osteoblasts that is critical for their effects on osteoclastogenesis. However, PGE2 augments osteoclast formation in spleen cell cultures stimulated by RANKL, indicating that PGE2 may stimulate osteoclastogenesis via receptors expressed on both osteoblasts/stromal cells and osteoclast progenitors (Wani et al., 1999; X Li et al., 2000). The importance of prostaglandin EP2 and EP4 receptors—not only for the osteoclastogenic effect of PGE2, but also for those of PTH and D3—has been demonstrated (X Li et al., 2000; Tomita et al., 2002).
IL-6 has been implicated as a cytokine involved in the stimulation of bone resorption in inflammatory conditions (e.g., rheumatoid arthritis, periodontitis), multiple myeloma, post-menopausal osteoporosis, and primary hyperparathyroidism. However, IL-6 stimulates neither osteoclast formation in mouse bone marrow cultures nor bone resorption in mouse calvariae (Tamura et al., 1993; Palmqvist et al., 2002). In contrast, other members of the IL-6 family of cytokines—such as IL-11, leukemia inhibitory factor (LIF), and oncostatin M (OSM)—stimulate osteoclastogenesis and bone resorption (Tamura et al., 1993; Palmqvist et al., 2002), demonstrating that it is not low expression of the signal-transducing protein gp130 that is the reason for the lack of effect by IL-6, but rather low expression of IL-6 receptors, shown to be true in mouse calvarial osteoblasts at the mRNA level (Palmqvist et al., 2002). However, when IL-6 is added to the soluble IL-6 receptor, both osteoclastogenesis and bone resorption are induced, and these effects can be inhibited by an antiserum neutralizing gp130. When spleen cells from wild-type mice are co-cultured with osteoblasts from transgenic mice constitutively expressing the human IL-6R, osteoclasts are formed in response to IL-6, even in the absence of soluble IL-6R (Udagawa et al., 1995). Since IL-6 does not stimulate osteoclastogenesis in spleen cell cultures from IL-6R transgenic mice, these findings also underscore the critical role of osteoblasts in osteoclast differentiation.
Although analysis of these data clearly indicates the importance of stromal cells/osteoblasts for osteoclastogenesis, it does not indicate by what molecular mechanisms osteoblasts control the development of osteoclasts. The observations in the osteopetrotic op/op mice of the role of M-CSF were the first to demonstrate the importance of an osteoblastic molecule for the formation of osteoclasts. The op/op mice exhibit an osteopetrotic phenotype due to lack of multinucleated osteoclasts. Co-cultures of spleen cells from op/op mice and wild-type osteoblasts are sensitive to the osteoclastogenic effect by D3, whereas co-cultures of spleen cells from wild-type mice and ob/ob osteoblasts are insensitive, indicating that the defect in the ob/ob mice is present in osteoblasts (Takahashi et al., 1991). The insensitivity of the latter co-cultures, as well as the in vivo phenotype, can be prevented by the addition of M-CSF (Felix et al., 1990; Komada et al., 1991; Takahashi et al., 1991). The demonstration in op/op mice of an extra thymidine insertion at base pair 262 in the coding region of the M-CSF gene, resulting in a TGA stop codon 21 base pairs downstream (Yoshida et al., 1990), clearly shows that the phenotype of op/op mice is due to functionally inactive M-CSF. Later it was shown that M-CSF is important for survival, proliferation, and differentiation of early osteoclast progenitor cells (Tanaka et al., 1993; Felix et al., 1994). However, M-CSF is a secreted molecule that interacts with its receptor c-fms expressed on osteoclast progenitor cells, and the critical role of this cytokine does not explain the requirement of cell-to-cell contact between stromal cells/osteoblasts and the osteoclast precursor cells. It was not until the very recent observations of the critical role of RANKL, expressed on the cell surfaces of stromal cells/osteoblasts, and its interaction with the receptor RANK, expressed on osteoclast progenitor cells, that the molecular mechanism involved in cell-to-cell contact was clarified. The activation of RANK by RANKL is inhibited by OPG, a soluble receptor with sequence homology to RANK, which is secreted by stromal cells/osteoblasts. The activation of RANK in osteoclast progenitor cells results in the commitment of the pool of M-CSF expanded precursors to differentiate into mature osteoclasts (see further below).
RANKL-RANK signaling is crucial not only for osteoclast progenitor cell differentiation but also for the activity of the mature, multinucleated osteoclasts (Fig. 2; Fuller et al., 1998).
(4) Receptor Activator of Nuclear Factor κB Ligand
By screening a cDNA library of a D3-stimulated mouse bone-marrow-derived stromal cell line ST-2 (previously shown to support osteoclastogenesis), Yasuda and co-workers (1998a) reported a clone encoding a 40-kDa protein and suggested it to represent the previously predicted osteoclast differentiating factor (ODF) expressed by stromal cells/osteoblasts. The same molecule was also cloned by Lacey et al. (1998) from an expression library of the murine myelomonocytic cell line 32D, and this molecule was called osteoprotegerin ligand (OPGL). Subsequently, it was shown that ODF/OPGL was identical to the previously discovered cytokine TNF-related activation-induced cytokine (TRANCE; Wong et al., 1997) and RANKL [In accordance with the guidelines by the American Society for Bone and Mineral Research President’s Committee on Nomenclature, this factor is denoted RANKL in the present review.] (Anderson et al., 1997).
The cloning of RANKL revealed it to be a member of the TNF ligand superfamily, a cytokine family that also includes TNF-α, TNF-β, CD40 ligand, Fas ligand, CD30 ligand, TWEAK, and TRAIL (Locksley et al., 2001). Like other members of the TNF-like family of cytokines, RANKL is a type II membrane-embedded protein, with a large extracellular, receptor-binding domain, a membrane-anchoring domain, and a connecting stalk. The RANKL gene is present on human chromosome 13q14 and on mouse chromosome 14. The mouse and human RANKL consists of 316 and 317 amino acid residues, respectively. Sequence alignments of the amino acid residues among proteins in the TNF ligand family show quite a low level of amino acid conservation between the members. However, information from x-ray crystal structures has demonstrated several common features of the three-dimensional structure. One feature of the ligands in the TNF-like family is that they form self-assembling, non-covalent homotrimers with β-pleated sheets assuming a ‘jellyroll’ orientation. The homology within the TNF ligand family is confined to domains involved in monomer folding and trimer assembly. The receptor-binding domains show only limited sequence homology between members of the TNF-like ligands, which is the basis for receptor selectivity. The shape of the ligand is that of an inverted bell, which at the base interacts with the receptors in 3:3 symmetric complex. Recently, the ectodomain of murine RANKL was crystallized, and it was shown that RANKL also self-associates as a homotrimer (Lam et al., 2001; Ito et al., 2002). The trimeric protein contains four unique surface loops that create the specificity in its interaction with the receptor RANK, elegantly demonstrated by site-directed mutagenesis of selected residues in these loops and functional osteoclastogenesis analysis with such mutated variants of recombinant RANKL in murine bone marrow cultures. Such structure-based comparative and functional studies will, in the future, be the basis for the rationale for and design of compounds affecting RANKL-induced osteoclastogenesis.
Like several other TNF-like type II proteins, RANKL trimers exist either as membrane-anchored proteins or in a soluble cleaved form, both being functionally active. Thus, the metalloprotease-disintegrin TNF-α convertase (TACE), or a related protease, can release the ectodomain of RANKL (Lum et al., 1999), and the metalloprotease inhibitor KB-R8301 inhibits the release of soluble RANKL (Nakashima et al., 2000).
RANKL is most abundantly expressed in trabecular bone, bone marrow, growth plate, periosteum, spleen, thymus, lymph nodes, and intestinal lymphoid patches. At the cellular level, high expression levels of RANKL can be found in several different stromal cells/osteoblasts and lymphoid cell lines. RANKL expression can also be found in hypertrophic chondrocytes.
The expression of RANKL by stromal cells and osteoblasts is regulated by a variety of hormones and cytokines that stimulate osteoclast formation and bone resorption. RANKL can be induced by PTH, PGE2, and forskolin, all acting via the cyclic AMP/protein kinase A (PKA) pathway, and by D3, acting via the VDR-mediated pathway (Lerner, 2000; Teitelbaum, 2000; Hofbauer and Heufelder, 2001; Takahashi et al., 2002). Cytokines in the IL-6 family of cytokines, including IL-6 (in the presence of soluble IL-6 receptor), IL-11, OSM, and LIF (Horwood et al., 1998; Ahlen et al., 2002; Palmqvist et al., 2002), as well as IL-7 (Toraldo et al., 2003) and IL-17 (Nakashima et al., 2000), also increase RANKL expression in osteoblasts and stromal cells. The stimulatory effect of IL-11 is potentiated by heparin (Walton et al., 2002). In addition, RANKL expression in osteoblasts can be induced by histamin (Deyama et al., 2002), IGF-I (Rubin et al., 2002a), or activation of purinergic P2Y receptors by ATP (Buckley et al., 2002), and decreased by melatonin (Koyama et al., 2002) or mechanical stress (Rubin et al., 2002b). The bone-resorptive effects of PTH, D3, and the IL-6 family of cytokines can be blocked by the inhibitory decoy receptor OPG (Palmqvist et al., 2002), thereby demonstrating the crucial role of increased RANKL expression for their bone-resorptive effects. In addition, IL-1β and TNF-α increase RANKL mRNA in human osteoblastic cells (Hofbauer et al., 1999a). Conditional expression of dominant-negative forms of different transcription factors has shown that the stimulatory effect of PTH on RANKL expression is dependent on cyclic AMP response element-binding protein (CREB), but not on c-Fos or Cbfa1 (Fu et al., 2002). Dominant-negative expression of STAT-3 or gp130 in the stromal cell line UAMS-32 suppresses the osteoclast-supporting activity of these cells stimulated by IL-6 (plus sIL-6R), OSM, or IL-11, but not stimulation caused by PTH or D3 (O’Brien et al., 1999), suggesting the importance of the gp130/STAT-3 signaling pathway in the regulation of RANKL gene expression. Ionomycin, thapsigargin, and phorbol 12-myristate 13-acetate (PMA) have also been shown to stimulate RANKL mRNA, demonstrating that the calcium/PKC pathway is also linked to RANKL expression (Takami et al., 2000). Not only the VDR, but also other steroid hormone receptors can induce RANKL, and such an effect has been demonstrated for the glucocorticoid receptor (Hofbauer et al., 1999c; Lerner et al., unpublished), the retinoid receptor (Lerner et al., unpublished), and the thyroid receptor (Miura et al., 2002). We have demonstrated that the neuropeptides vasoactive intestinal peptide (VIP) and pituitary adenylate cyclase activating peptide (PACAP) inhibit osteoclastogenesis induced by PTH and D3, and that this effect is associated with decreased RANKL mRNA expression (Mukohyama et al., 2000).
The promoter region of the RANKL gene contains a response element for core-binding factor a1 (Cbfa-1; also known as AML-3, Pebp2aA, or Runx2), which is a transcription factor crucial for osteoblast differentiation and expression of bone matrix proteins (Ducy et al., 1997). Consequently, cbfa-1 −/− mice have reduced mRNA expression of RANKL (Gao et al., 1998). In the periosteum of these mice, a few TRAP-positive osteoclasts can be seen, but they are smaller and have a reduced number of nuclei (Komori et al., 1997). In line with these observations, osteoblasts from cbfa-1 −/− mice are less effective in supporting osteoclast formation when co-cultured with spleen cells from wild-type mice (Gao et al., 1998). Although cbfa-1 does not stimulate the 0.7-kb 5′-flanking region of the RANKL gene containing two putative binding sites for cbfa-1 (O’Brien et al., 2002), it was recently shown that adenoviral introduction of cbfa-1 into CA.120-4 cells induced RANKL expression (Enomoto et al., 2003). However, forced expression of RANKL in cbfa-1 −/− mice did not completely restore osteoclast formation, suggesting that additional factors are missing in cbfa-1 knockout mice. The role of cbfa-1 in osteoclastogenesis was also demonstrated by the observation that transgenic mice overexpressing cbfa-1 under the control of the collagen α(1) type I collagen promoter have a high bone turnover rate due to increased bone resorption and bone formation (Geoffroy et al., 2002). This phenotype is associated with increased formation of osteoclasts in bone marrow cultures treated with D3, as well as an increased number of osteoclasts in D3-stimulated co-cultures of transgenic osteoblasts with spleen cells from either wild-type or transgenic mice. The detailed knowledge of RANKL gene induction, however, has to await further analysis of the RANKL gene promoter region.
The crucial role of RANKL in osteoclastogenesis has been demonstrated in mice deficient in RANKL due to targeted deletion of the RANKL gene (Kong et al., 1999a). These mice exhibit a severe form of osteopetrosis due to complete absence of osteoclasts. Similar to many other forms of osteopetrosis, RANKL −/− mice have impaired tooth eruption. The lack of RANKL also results in disturbances in the growth plate, and the bones are shorter than normal. The osteopetrotic phenotype cannot be restored by bone marrow transplantation, which indicates that the lack of osteoclasts is not due to defective hematopoietic cells, a view which is in line with the fact that RANKL is expressed in stromal cells/osteoblasts. Although no TRAP-positive osteoclast progenitor cells can be seen in RANKL −/− mice, these mice have normal osteoclast progenitor cells, as demonstrated by the observation that osteoclast progenitor cells from RANKL −/− can differentiate to mature osteoclasts when co-cultured with osteoblasts from wild-type mice. In contrast, mature osteoclasts are not formed when osteoblasts from RANKL −/− are co-cultured with osteoclast precursors from wild-type mice (Kong et al., 1999a). Interestingly, in view of the very close relationship among osteoclasts, macrophages, and dendritic cells, the differentiation and function of macrophages and dendritic cells seem to be normal in RANKL-deficient mice. Similar to many osteopetrotic animals, RANKL −/− mice also have splenomegaly due to extramedullary hematopoiesis.
Mice overexpressing soluble RANKL under the control of the liver-specific human serum amyloid P component promoter exhibit increased numbers of osteoclasts and decreased bone mineral density (Mizuno et al., 2002). When soluble RANKL was expressed ubiquitously under the control of the β-actin promoter, the mice died at the late fetal stage. Administration of RANKL for 7 days to rats also results in decreased bone mineral density, an effect observed in both cortical and trabecular bone (McHugh et al., 2003).
Besides the osteopetrotic phenotype, RANKL −/− mice exhibit impaired early T- and B-lymphocyte differentiation and a complete lack of lymph nodes (Kong et al., 1999a), which is likely to be related to RANKL/TRANCE expression in lymph nodes, spleen, thymus, and intestinal lymphoid patches (Wong et al., 1997; Lacey et al., 1998). However, spleen and Peyer’s patches seem to be normal. RANKL can be induced in mammary epithelial cells by injection of progesterone, prolactin, and parathyroid-hormone-related peptide (PTHrP), but not by estrogen (Fata et al., 2000). The regulation of RANKL by the pregnancy hormones indicates a role for this cytokine in mammary gland development required for lactation. In line with this hypothesis, Fata et al. (2000) also reported that RANKL −/− female mice fail to form lobulo-alveolar mammary structures during pregnancy, leading to the death of newborn pups.
(5) Receptor Activator of Nuclear Factor κB
RANK was initially cloned from a cDNA library of human dendritic cells (Anderson et al., 1997). Later, RANK was demonstrated to be the receptor for OPGL/RANKL on osteoclasts (Hsu et al., 1999). Like other members of the TNF-R superfamily, RANK is a type I transmembrane protein containing four cysteine-rich pseudorepeat domains in the extracellular region, a hallmark of the TNF-R family (Locksley et al., 2001). The mouse and human RANKs contain 625 and 616 amino acid residues, respectively, the latter having a signal peptide (28 amino acids), an N-terminal extracellular domain (184 amino acids), a transmembrane spanning domain (21 amino acids), and a large C-terminal cytoplasmic tail (383 amino acids). In the human genome, RANK is present on chromosome 18q22.1. The extracellular part of TNF-R-like receptors forms elongated structures by a scaffold of disulfide bridges, which fit into the ‘inverted bell/groove’ of the ligand trimer in a 3:3 complex (Locksley et al., 2001). This has recently been demonstrated to be true also for the interaction between RANKL and RANK (Lam et al., 2001). The intracellular tails of the receptor form a 3:3 complex with signaling proteins like TRAFs and FAS-associated with death domain (FADD) proteins (see further below). The reason for this stoichiometry with the three-fold symmetry for both ligand-binding and signaling complex formation remains an evolutionarily interesting issue. The requirement of RANK clustering is also indicated by the finding that polyclonal antibodies recognizing the extracellular domain of RANK can stimulate osteoclast formation in vitro (Nakagawa et al., 1998; Hsu et al., 1999).
RANK can be demonstrated at the mRNA level in many organs and tissues, but at the cellular level it is mainly expressed in osteoclast progenitor cells, osteoclasts, B- and T-lymphocytes, and in dendritic cells (Anderson et al., 1997; Hsu et al., 1999). Regulation of RANK expression has been much less extensively studied than that of RANKL. It has been reported, however, that TGF-β (Yan et al., 2001) and D3 (Ahlen et al., 2002; Palmqvist et al., 2002) can increase RANK mRNA and that IL-4 (Lerner and Conaway, 2000) as well as activation of gp130 by IL-6 (and the sIL-6R; Palmqvist et al., 2002) can decrease RANK mRNA. The effects of TGF-β and D3 are in accordance with their osteoclastogenic effects, which also is true for IL-4, a known inhibitor of osteoclast formation. However, the decreased RANK expression caused by gp130 stimulation is surprising when one considers the stimulatory effect on bone resorption by IL-6 (+ sIL-6R). The inhibitory effect of IL-4 on RANK expression in osteoclast progenitor cells is in contrast to the stimulatory effect on T-lymphocytes (Anderson et al., 1997). Recently, we have found that dexamethasone also enhances the mRNA expression of RANK, which may partly explain the potentiation by glucocorticoids on osteoclast formation induced by PTH and D3. Interestingly, neither TNF nor RANKL affects RANK expression, but co-stimulation with both cytokines results in a substantial up-regulation of RANK mRNA (Zhang et al., 2001), which is likely to be one mechanism by which TNF synergistically potentiates the osteoclastogenic effect of RANKL (Abu-Amer et al., 2000; Lam et al., 2000; Zhang et al., 2001). The neuropeptides VIP and PACAP-38 have been shown to decrease D3-stimulated RANK mRNA, which might, at least partly, explain the inhibitory effects of these neuropeptides on osteoclastogenesis (Mukohyama et al., 2000).
Similar to RANKL-deficient mice, RANK knockout mice exhibit an osteopetrotic phenotype due to the absence of multinucleated osteoclasts (Dougall et al., 1999; J Li et al., 2000). The bone marrow cavities are occluded and filled with cartilage encased in mineralized bone matrix. Similar to RANKL −/− mice, the mice deficient in RANK exhibit widened and disturbed organization of the growth plate. The osteopetrotic phenotype of RANK −/− mice, in contrast to that of RANKL −/− mice, can be prevented by bone marrow transplantation, demonstrating the intrinsic defect in the osteoclastic lineage of RANK −/− mice. This is also demonstrated by the fact that no osteoclasts are formed when spleen cells are incubated with M-CSF and RANKL. The absence of osteoclasts in RANK −/− mice is not due to a lack of myeloid or progenitor stem cells, since differentiation and function of macrophages and dendritic cells from their myeloid precursors are normal. The RANK-deficient mice exhibit increased extramedullary hematopoiesis in the spleen, but not in the liver, and a deficiency of B-cells in the spleen. The teeth in RANK −/− mice are smaller, with apparently normal enamel, dentin, cementum, and odontoblasts, but fail to erupt, the latter being a typical finding in mice with decreased osteoclast differentiation or function. RANK deficiency also results in hypocalcemia, hypophosphatemia, and elevated serum levels of PTH. These mice are also resistant to the hypercalcemic response and to the increase of osteoclast number induced by injection of PTHrP, D3, or IL-1β. Nor does TNF-α cause any hypercalcemia. Interestingly, however, osteoclasts are formed in the vicinity of the site of TNF-α injection, suggesting that TNF-α signaling can partly compensate for the lack of RANKL-RANK interaction. The RANK −/− mice have normal intestinal lymphoid tissues, including Peyer’s patches, but completely lack all other peripheral lymph nodes. Thus, the phenotype of RANK −/− mice is identical to that of RANKL −/− mice, with the exception that thymic differentiation is intact in RANK −/− mice but impaired in RANKL −/− mice.
(6) Osteoprotegerin
Osteoprotegerin was initially discovered as an ‘osteoclast-inhibitory factor’ (OCIF) released as a heparin-binding protein from human skin fibroblasts and found to inhibit osteoclast formation in vitro (Tsuda et al., 1997). OPG was cloned in a fetal rat intestine cDNA-sequencing project and given the name osteoprotegerin (Simonet et al., 1997). OCIF was then cloned and found to be identical to OPG (Yasuda et al., 1998b). OPG is also identical to TR-1, which was identified in a search of an expressed sequence tag database (Kwon et al., 1998), and to follicular dendritic receptor 1 (FDCR-1; Yun et al., 1998). OPG is synthesized in humans, rats, and mice as a 401-amino-acid protein, which, after cleavage of a 21-amino-acid signal peptide, results in a 380-amino-acid mature protein. In humans, the OPG gene is located on chromosome 8q23-24. The amino acid sequence of OPG displays several homologies to members of the TNF-R superfamily, including RANK. Human, rat, and mouse OPG has large homologies (85–95%). The 60-kDa OPG protein exists as a monomer which forms 120-kDa disulfide-linked homodimers containing several N-glycosylation sites. OPG contains four cysteine-rich domains in the N-terminal end, two homologous ‘death domains’ (DDH) in the C-terminus, a heparin-binding site, and a cysteine residue required for homodimerization but, in contrast to other members of the TNF-R superfamily, lacks a transmembrane spanning domain and a cytoplasmic tail. These features make OPG a unique protein in the TNF-R superfamily, and, unlike the other members, it is a secreted protein. The biological effect of OPG is dependent on the integrity of the four cysteine-rich domains. The DDH regions of OPG are similar to the ‘death domains’ of TNF-R1 and FAS, which mediate apoptotic signals. The function of the DDH region of OPG, however, is elusive but is not involved in the inhibitory effect on osteoclast formation and function.
Secreted OPG acts as a ‘decoy receptor’ due to its affinity to both membrane-bound and soluble RANKL and prevents the activation of RANK. Because OPG is present as a homodimer and because of the low degree of sequence homology between RANK and OPG, it has been questioned if OPG binds to RANKL in a manner similar to the trimeric interaction between RANK and RANKL. OPG can also bind to TRAIL, a member of the TNF ligand superfamily, although the affinity between OPG and TRAIL is considerably less than that between OPG and RANKL.
Because of its ‘decoy receptor’ function, OPG inhibits osteoclast formation in bone marrow cultures, as well as bone resorption in organ-cultured fetal rat long bones and newborn murine calvariae stimulated by a variety of hormones and cytokines (Kwon et al., 1998; Palmqvist et al., 2002). Injection of OPG into rats or mice results in decreased bone mineral density, bone volume, trabecular bone area, and reduced numbers of osteoclasts (Simonet et al., 1997; Yasuda et al., 1998a). Transgenic mice overexpressing OPG, under the control of the apolipoprotein E gene promoter and its liver-specific enhancer, have a normal appearance, but the bones are osteopetrotic with enhanced bone mineral density, primarily in the trabecular bone, and have decreased numbers of trabecular osteoclasts (Simonet et al., 1997). In contrast to RANKL −/− and RANK −/− mice, the shapes and sizes of bones are normal, and no abnormalities in tooth eruption, lymphocyte development, or thymus and lymph node organogenesis can be found. However, similar to most osteopetrotic animals, OPG transgenic mice exhibit splenomegaly. The decrease of osteoclasts is not associated with any decrease of F4/80-positive monocyte/osteoclast progenitor cells.
Targeted ablation of the OPG gene results in mice exhibiting a substantial decrease of bone density in both trabecular and cortical bone (Bucay et al., 1998; Mizuno et al., 1998). This phenotype is evident early post-natally, and an increased incidence of vertebral and endochondral bone fractures was already noted within the first two weeks. Thus, OPG −/− mice show signs of early-onset osteoporosis. Histological analysis revealed that the bone present in OPG-deficient mice is of mainly a woven type, reflecting decreased remodeling of the skeleton. The decreased bone density is due to enhanced numbers of osteoclasts in the skeleton. The finding that osteoclast formation in spleen cell and bone marrow cultures stimulated by RANKL and M-CSF is similar in OPG −/− mice and wild-type mice indicates that the intrinsic defect is not confined to the number of osteoclast progenitor cells and is in agreement with the finding that OPG is expressed by stromal cells/osteoblasts. This view is further supported by the observations that: (i) osteoblasts from OPG −/− mice support osteoclast formation in co-cultures with osteoclast progenitor cells, even in the absence of hormones stimulating osteoclastogenesis; (ii) basal bone resorption in cultured fetal rat long bones is increased when compared with resorption in wild-type mice; and (iii) RANKL mRNA expression in osteoblasts from OPG −/− is not different from that in wild-type mice (Udagawa et al., 2000). Surprisingly, OPG-deficient mice also exhibit calcification in the media of aorta and renal arteries, but not in smaller arteries, veins, or capillaries.
OPG is expressed more widely and to a much higher extent than RANKL. OPG mRNA has been detected in bone, cartilage, aorta, skin, lung, heart, kidney, liver, brain, and in several other tissues. At the cellular level, OPG is expressed in osteoblasts, stromal cells, endothelial cells, aortic smooth-muscle cells, fibroblasts, dendritic cells, and lymphoid cell lines. It is apparent that osteoclast formation and activation are critically regulated by the RANKL-RANK-OPG system and that the relative expression of these molecules will determine the numbers of osteoclasts formed and consequently the bone mineral density of the skeleton. Very potent stimulators of osteoclastogenesis and bone resorption, such as PTH and D3, substantially increase the ratio RANKL/OPG by increasing RANKL and decreasing OPG expression. At variance, stimulators of bone resorption—such as IL-1, TNF-α, TGF-β, IL-6 (+sIL-6R), IL-11, OSM, and LIF—increase both RANKL and OPG expression, which is the likely explanation for the reduced effectiveness of these cytokines to stimulate bone resorption as compared with PTH and D3. Glucocorticoids, which stimulate bone resorption in organ-cultured bones and synergistically potentiate the osteoclastogenic effects of PTH and D3 in bone marrow cultures, decrease OPG expression in osteoblasts. The suppressing effect of PTH on OPG expression is mimicked by forskolin (Takami et al., 2000) and is at variance with PTH-induced RANKL expression, which is dependent on both the transcription factors CREB and c-fos, but not on Cbfa1 (Fu et al., 2002). Activation of PKC does not mimic the stimulatory effect of PTH but leads to increased OPG expression (Takami et al., 2000).
The relatively restricted phenotype of OPG transgenic and knockout mice is somewhat surprising when one considers the ubiquitous expression of OPG and the fact that RANKL expression is not restricted only to bone (Simonet et al., 1997). This suggests that OPG treatment of pathologically increased bone resorption would be an interesting possibility. Clinical trials are already ongoing for the treatment of post-menopausal osteoporosis and metastatic bone disease.
The importance of OPG has also been investigated by studies of the presence and distribution of polymorphisms in promoter and intron regions of the OPG gene (Langdahl et al., 2002). Twelve different polymorphisms were detected; none of these was associated with changes in bone mineral density or biochemical markers of bone turnover in normal controls. Two of the alleles, A163G and T245C, were significantly more common in osteoporotic patients with vertebral fractures.
(7) Downstream Intracellular Signaling Molecules
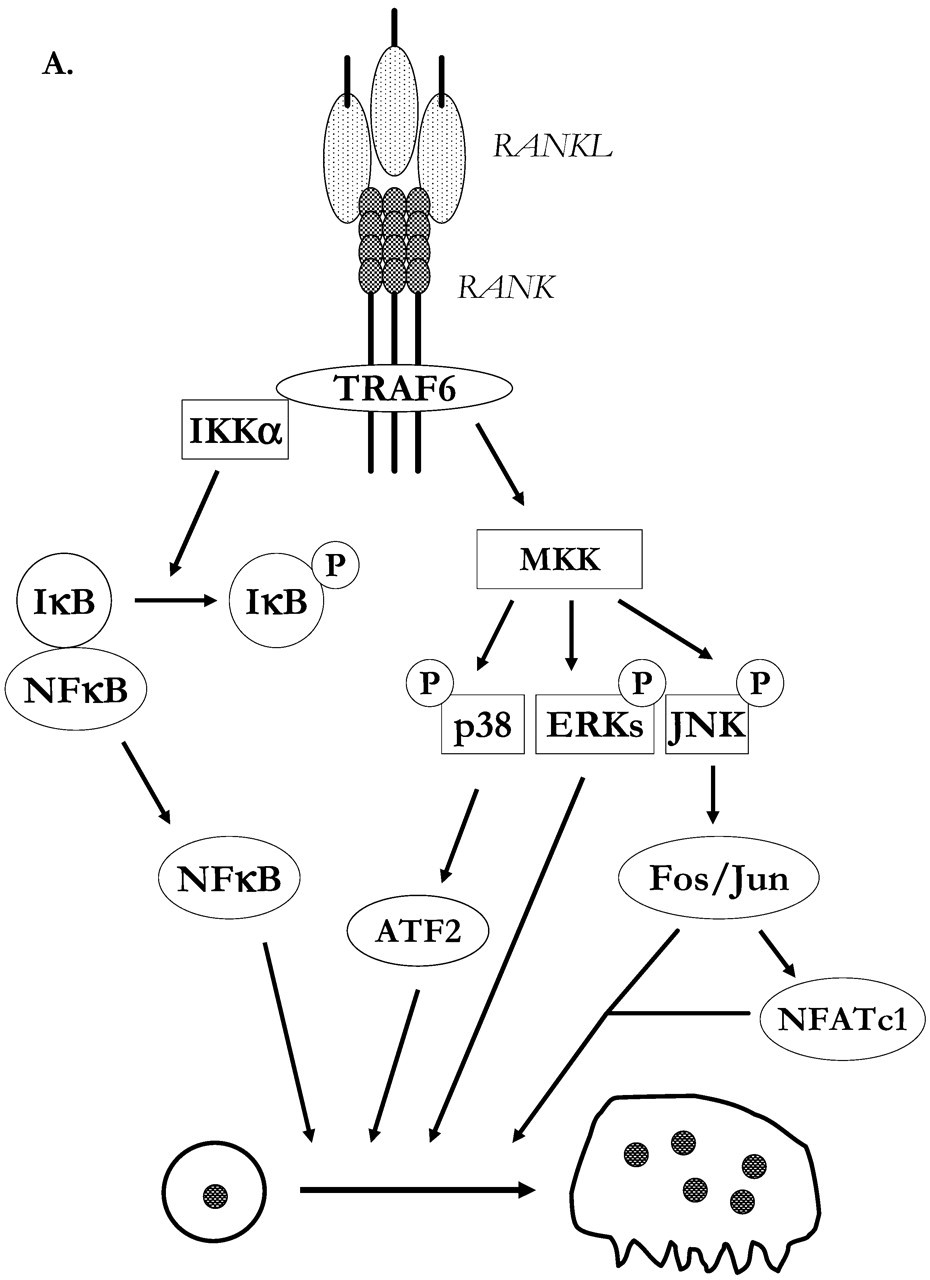
TRAF family members are cytoplasmic adapter proteins which are recruited by receptors of the TNF-R superfamily and the Toll/IL-1 receptor family. The TRAF domain is located in the C-terminal region of all members of the TRAF family. The trimeric cytoplasmic tails of clustered RANK interact in the membrane-proximal domain with TRAF6 at a binding site that is distinct from those binding TRAF1, TRAF2, TRAF3, and TRAF5. TRAF6 is distinct from the other TRAFs, since it is the only one involved in Toll/IL-1 receptor signaling. The activation of TRAF6, similar to the activation of TFAF2 and TRAF5, results in stimulation of transcription factors AP-1 and NF-κB through activation of MAPKs and inhibitory κB kinase (IKK), respectively (Fig. 3A). Two groups have generated TRAF6 −/− mice and found them to be osteopetrotic and to exhibit defective tooth eruption, B-cell differentiation, lymph node organogenesis, and IL-1 signaling (Lomaga et al., 1999; Naito et al., 1999). In the study by Lomaga et al. (1999), the osteopetrotic mice had normal amounts of TRAP-positive multinucleated osteoclasts, but the osteoclasts lacked contact with bone surfaces, indicating that it was the later stages of osteoclast differentiation, including polarization and activation, rather than differentiation and fusion of osteoclast progenitor cells, that are dependent on TRAF6 expression. This is, to some extent, unexpected, since deficiency of RANK leads to arrest of osteoclast progenitor cell differentiation. Similar to the phenotype of RANK −/− mice, the TRAF6 −/− mice generated by Naito et al. (1999) lacked TRAP-positive multinucleated osteoclasts, and the osteoclast progenitor cells do not respond to RANKL/M-CSF stimulation with increased formation of mature osteoclasts. The discrepancy in the two TRAF6 −/− models may be due to differences in targeting constructs, indicating the possibility that different domains of TRAF6 might be important for differentiation and maturation of osteoclasts. Transfection of TRAF6 −/− spleen cells with wild-type TRAF6 restores the osteoclastogenic response to RANKL/M-CSF (Kobayashi et al., 2001). Using different constructs, Kobayashi et al. (2001) were able to demonstrate that the RING finger of TRAF6 is responsible for maturation of osteoclasts, whereas the second and third zinc fingers are important for differentiation of the osteoclast progenitor cells. Thus, different subdomains of TRAF6 seem to be involved in distinct post-receptor pathways. In line with this view, Kobayashi et al. (2001) demonstrated that the RING finger is necessary for full activation of c-Jun amino-terminal kinase (JNK) and p38 MAPK, but not for NF-κB activation. Further support for the view of the presence of different functional domains in the interactions between RANK and TRAFs has been presented by Armstrong et al. (2002), using spleen cells from RANK −/− mice transfected with different RANK constructs selectively incapable of binding different TRAF proteins. It was shown that osteoclast progenitor cell differentiation to multinucleated osteoclasts was not blocked unless all TRAF-binding sites were deleted. In contrast to this functional redundancy in the TRAF pathways downstream from RANK with regard to differentiation, it was demonstrated that TRAF6 is indispensable for the organization of the osteoclast cytoskeleton and the resorptive activity, in line with the observations by Lomaga et al. (1999) in TRAF6 −/− mice. The osteopetrotic phenotype of TRAF6 −/− mice is not observed in TRAF2- or TRAF5-deficient mice. However, TRAF5 −/− mice still exhibit a bone cell phenotype, since osteoclast formation in bone marrow cultures stimulated with M-CSF and either RANKL or TNF-α is significantly decreased (Kanazawa et al., 2003). Deletion of the TRAF5 gene also results in a delayed hypercalcemic response in mice injected with PTH.
Activation of osteoclast progenitor cells with RANKL/RANK/TRAF6 initiates a cascade of kinases (Fig. 3A). Thus, RANK stimulation causes activation of IKKα, which induces a rapid serine phosphorylation of IκB, leading to ubiquitination and proteasomal degradation (Wei et al., 2001). This is followed by transactivation of the IκB gene and increased cytosolic IκB protein. As a consequence of IκB phosphorylation, the IκB/NF-κB complex dissociates, followed by translocation of the NF-κB subunits p50/p65 to the nucleus. NF-κB consists of homo- or heterodimers of five members of the Rel family, including p50 (NF-κB1), p52 (NF-κB2), p65 (RelA), RelB, and c-Rel. The importance of the NF-κB pathway for osteoclast formation is demonstrated by the finding that mice deficient in both NF-κB subunits p50 and p52 are osteopetrotic, with marrow cavities filled with unremodeled osteocartilaginous matrix (Franzoso et al., 1997; Iotsova et al., 1997). The p50/p52 −/− mice lack not only mature osteoclasts but also TRAP-positive mononuclear progenitor cells. However, RANK-expressing osteoclast progenitors are present in p50/p52 knockouts (Xing et al., 2002). That it is the lack of p50/p52 expression in osteoclast progenitor cells that is the cause of defective osteoclast formation is indicated by the observations that co-culture of osteoblasts from knockout mice with spleen cells from wild-type mice results in the formation of mature osteoclasts, whereas co-culture of wild-type (or knockout) osteoblasts with spleen cells from p50/p52-deficient mice does not result in the formation of osteoclasts. In line with these observations, the osteopetrotic phenotype of p50/p52 −/− mice can be prevented by bone marrow transplantation. Activation of NF-κB by RANK is dependent on RANK-induced elevations of intracellular Ca2+ (Komarova et al., 2003).
As described above, the transcription factor c-Fos signaling is an important pathway downstream RANK (Grigoriadis et al., 1994). In a genome-wide screening of mRNAs expressed in RANKL-stimulated RAW 264.7 cells (Ishida et al., 2002) and in RANKL-stimulated mouse bone marrow cells (Takayanagi et al., 2002b), it has been shown that the transcription factor NFATc1 (=NFAT2/NFATc) is one of the strongest genes induced. NFATc1 is a member of the nuclear factor of activated T-cells (NFAT) family of transcription factors, which were originally found to be important in regulation of the immune system. Induction of NFATc1 involves RANK-induced Ca2+ oscillations and activation of Ca2+/calmodulin-dependent calcineurin (a serine/threonine phosphatase), which is necessary for translocation of NFATc1 to the nucleus (Takayanagi et al., 2002b). Loss-of-function mutation in the NFATc1 gene leads to abolished capacity to form osteoclasts after RANKL stimulation, whereas M-CSF stimulation of monocyte/macrophage precursors is normal (Takayanagi et al., 2002b). NFATc1 forms a complex with c-Fos, and expression of both transcription factors is necessary for the expression of TRAP, and calcitonin receptors, which are similar to the genes for cathepsin K, carbonic anhydrase II, and MMP-9, contain binding sites for both NFATc1 and AP-1.
Three major subfamilies of MAP kinases have been identified: (i) extracellular signal-regulated kinases (ERKs), (ii) JNK, and (iii) p38 MAP kinase. These can be activated by MAP kinase kinase (MKK)-mediated phosphorylation on threonine and tyrosine residues. The involvement of this pathway in osteoclastogenesis is indicated by the observation that JNK is not activated in TRAF6 −/− mice (Lomaga et al., 1999) and by the finding that RANK overexpression leads to enhanced activation of JNK and NF-κB (Hsu et al., 1999). Using cells transfected with RANK C-terminal deletion mutants, Hsu et al. (1999) showed that RANK-induced activation of JNK and NF-κB correlates with the TRAF6-binding domain of RANK. Matsumoto et al. (2000) have shown that RANKL stimulates phosphorylation of JNK, ERK, and p38 MAP kinase in RAW 264.7 cells, an osteoclast progenitor cell line which can differentiate to mature osteoclasts in the presence of RANKL. Pharmacological inhibitors have been used to show that osteoclastogenesis was associated with p38 MAP kinase, and strong evidence for the crucial role of this kinase was provided by demonstrations that the expression of the dominant-negative form of either p38 MAP kinase or MKK6 inhibited RANKL-induced osteoclast formation. RANKL-induced stimulation of p38 MAPK is involved in the differentiation of osteoclasts, but not in their survival or bone-resorbing activity (Li et al., 2002). Recently, David et al. (2002) have shown, by using JNK1 −/− and JNK2 −/− mice, that RANKL activates JNK1 preferentially and that lack of JNK1 results in decreased (~ 50%) osteoclast formation. In addition, RANKL stimulation of bone marrow cells from either c-Jun −/− or JunD −/− mice, or RANKL-stimulated bone marrow cultures from mice (JunAA/JunAA) carrying a c-Jun mutant rendering c-Jun less sensitive to phosphorylation by JNK, revealed that JNK-1-dependent phosphorylation of c-Jun, and c-Jun itself, are important for RANKL-induced osteoclastogenesis. Interestingly, JNK1 seems to affect osteoclastogenesis by two mechanisms: (i) JNK1 protects osteoclast progenitor cells from RANKL-induced apoptosis, a mechanism independent of c-Jun phosphorylation; and (ii) a c-Jun phosphorylation-dependent increased differentiation of osteoclast progenitor cells.
Signaling pathways downstream of RANK (similar to that of M-CSF) also include activation of PI3K and of the anti-apoptotic serine/threonine kinase Akt (protein kinase B) (Fig. 3B). Activation of PI3K and Akt by RANK is dependent on TRAF6/c-src interactions (Wong et al., 1999; Arron et al., 2001). The Akt survival signaling pathway is crucial for the anti-apoptotic and osteoclastogenic activity of RANK. Recently, Sugatani et al. (2003) reported that RANK activation of Akt results in phosphorylation of Bad, a member of the Bcl-2 family, which, through phosphorylation of Ser-136 by Akt, becomes inactivated and loses its apoptotic activity. The PI3K/Akt/Bad pathway is inhibited by the tumor suppressor gene PTEN (phosphatase and tensin homologue deleted from chromosome 10; Sugatani et al., 2003). PI3K generates the lipid second messenger phosphatidylinositol 3,4,5-trisphosphate [PtdIns(3,4,5)P3], which can be dephosphorylated by PTEN to the inactive PtdIns(4,5)P2. PTEN also can act as a protein phosphatase by dephosphorylating active Akt. Thus, PTEN can inhibit the PI3K/Akt pathway by two mechanisms. PTEN overexpression suppresses RANKL-stimulated differentiation of RAW 264.7 cells to osteoclasts, whereas transfection with dominant-negative PTEN increases osteoclast formation. Although the data cannot fully discriminate between effects of PTEN on osteoclast number as being a consequence of effects on differentiation or on cell survival/apoptosis, the most likely explanation is that PTEN interferes with the PI3K/Akt/Bad signaling involved in RANK-mediated anti-apoptosis. Sugatani et al. (2003) also provide evidence that RANK may regulate the expression of PTEN in osteoclasts, which suggests that PTEN plays a role in regulating the balance between active and non-active Akt. Interestingly, inhibition of PI3K by PTEN also inhibits osteopontin-stimulated migration of RAW 264.7 cells, by a mechanism unrelated to Akt/Bad signaling. Besides PTEN [dephosphorylating PtdIns(3,4,5)P3 by acting as a 3′-phosphatase], SHIP and SHIP2 can also contribute to the dephosphorylation of PtdIns(3,4,5)P3 by acting as 5′-phosphatases. SHIP is a hematopoietic-restricted Src homology (SH) 2-containing inositol-5-phosphatase (SHIP), which is tyrosine-phosphorylated by various cytokines, including M-CSF. Mice deficient in SHIP have increased numbers of osteoclasts due to the increased life span of these cells and to increased sensitivity to the osteoclastogenic effects of M-CSF and RANKL (Takeshita et al., 2002). SHIP −/− osteoclasts are enlarged and contain huge numbers of nuclei. Due to the large numbers of giant osteoclasts, mice deficient in SHIP are osteoporotic with decreased bone mass and reduced trabecular thickness, number, and connectivity. Thus, inhibition of the PI3K pathway, by either PTEN or SHIP, seems to be a negative regulator of osteoclast formation. The phenotype of SHIP −/− mice, including large osteoclasts containing a huge number of nuclei, increased serum levels of IL-6, and hypersensitivity to RANKL, is very similar to that of patients with Paget’s disease.
(8) RANKL-RANK-OPG in Dental Tissues
In dental cells, OPG and RANKL mRNA and protein have been found to be expressed in periodontal ligament cells, pulp cells, odontoblasts and in follicle cells (Sakata et al., 1999; Rani and MacDougall, 2000; Wise et al., 2000; Wada et al., 2001; Hasegawa et al., 2002a). Similar to previous observations in osteoblasts, RANKL mRNA is increased in human periodontal ligament cells by D3, whereas OPG mRNA is decreased (Hasegawa et al., 2002b). M-CSF has been detected in rat follicle cells, odontoblasts, and pulp cells (Rani and MacDougall, 2000; Wise et al., 2002a), and recently, human gingival cells have been found to constitutively express large amounts of M-CSF mRNA (Lerner et al., unpublished). In addition, RANKL and OPG have been found in ameloblasts (Rani and MacDougall, 2000). RANK has been found to be expressed in mononuclear cells presumed to be osteoclast progenitor cells and in osteoclasts/odontoclasts observed in sites with root resorption of human deciduous teeth (Lossdörfer et al., 2002). This means that the key molecules in osteoclast progenitor cell expansion and differentiation are expressed both in the pulp tissue and in the follicle cells. It is likely that regulation of the expression of M-CSF, RANKL, and OPG is important for the control of osteoclast/odontoclast formation within the pulp tissue and the periodontal ligament, processes seen in root resorption and tooth eruption. Interestingly, the expression of OPG during normal conditions seems to be considerably greater than that of RANKL in periodontal ligament cells and in dental follicle cells, resulting in cells that inhibit osteoclast formation in spleen cell cultures, an effect that can be blocked by antibodies neutralizing OPG (Kanzaki et al., 2001; Wise et al., 2002a). During the initial phases of tooth eruption, at the time point when osteoclast progenitor cells are accumulated in the vicinity of the tooth bud crown, OPG expression in the dental follicle is down-regulated (Wise et al., 2000).
The crucial role of the RANKL-RANK-OPG system in tooth eruption is most obvious by the observations that mice deficient in RANKL or RANK (or some of the downstream signaling molecules) do not have erupted teeth. For an extensive review of the molecular mechanisms involved in tooth eruption, the reader is referred to Wise et al. (2002b).
The RANKL-RANK-OPG system is also involved in the bone resorption taking place during orthodontic tooth movement. In wild-type mice, osteoclasts are formed at the pressure site in the periodontal ligament and in adjacent alveolar bone. In OPG −/− mice, the number of osteoclasts formed increases dramatically, leading to extensive resorption and perforation of alveolar bone as early as at 2 and 5 days after force application (Oshiro et al., 2002).
(9) RANKL/RANK/OPG System in Pathological Conditions
Excessive osteoclastic resorption is a common feature of chronic inflammatory processes, e.g., rheumatoid arthritis, loosened joint prosthesis, and periodontitis. Several lines of evidence point to an important role for RANK activation in these diseases. In physiological bone remodeling, the cell-to-cell contact between RANKL-expressing stromal cells/osteoblasts and RANK-expressing monocyte/osteoclast precursor cells is crucial. In inflammatory processes, activated T-lymphocytes express RANKL, and it is therefore possible that cell-to-cell contact between T-lymphocytes and monocytes/osteoclast precursors is involved in osteoclast formation. It was initially demonstrated that antigen activation of CD8+ and CD4+ T-cells resulted in expression of RANKL and that co-culture of such cells with osteoclast progenitor cells gave rise to formation of mature osteoclasts by a mechanism sensitive to inhibition by OPG (Kong et al., 1999b). Further support for the involvement of RANKL-expressing T-cells in osteoclast formation is provided by the finding that Ctla4 −/− mice, with spontaneously activated T-cells, exhibit osteoporosis and that daily injections of OPG decrease osteoclast development and increase bone mineral density (Kong et al., 1999b).
B-lymphocytes may also participate in osteoclast formation, either by expressing RANKL or by serving, themselves, as osteoclast progenitor cells (Manabe et al., 2001). Interestingly, disruption of the klotho gene results in mice with osteopetrosis resembling senile osteopetrosis, and the decreased number of osteoclasts in these mice is associated with a reduced number of B-cells (Manabe et al., 2001). Excessive expression of RANKL by B-cells may play an important role in the bone loss seen in patients with multiple myeloma (Terpos et al., 2003).
OPG treatment also decreases local bone loss and osteoclast number in rats with experimentally induced adjuvant arthritis (Kong et al., 1999b) and in rats with collagen-induced arthritis (Romas et al., 2002a). RANKL-expressing lymphocytes have also been demonstrated in synovium from patients with rheumatoid arthritis (Haynes et al., 2003). OPG has been demonstrated in endothelial cells and in the synovial lining cells in patients with synovitis, except in those with active rheumatoid arthritis. The importance of the RANKL-RANK-OPG system in rheumatoid arthritis has recently been reviewed by several groups (Jones et al., 2002; Romas et al., 2002b; Theill et al., 2002; Udagawa et al., 2002).
RANKL-RANK signaling has also been implicated in wear-debris-induced bone resorption and loosening of joint prostheses. Thus, RANKL expression and osteoclast formation in the bone marrow of wild-type mice can be induced by polymethylmethacrylate particles (Clohisy et al., 2003). Similarly, bone resorption in mouse calvariae caused by implanted titanium can be blocked by antibodies neutralizing RANK and is absent in calvariae of RANK −/− mice (Childs et al., 2002).
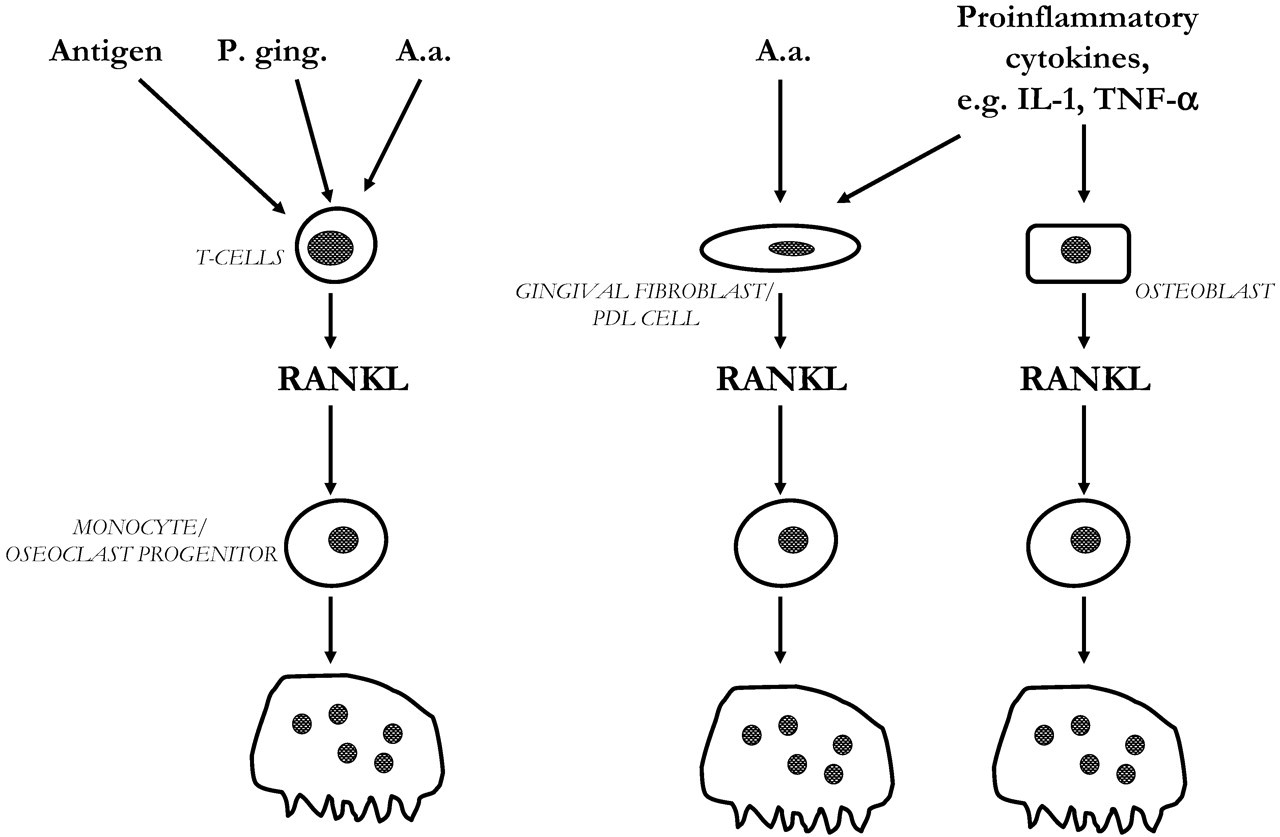
In periodontal disease, dense infiltrates of mononuclear leukocytes are found in the gingiva, including CD4+ and CD8+ T-cells, and moncytes/osteoclast progenitor cells (Taubman and Kawai, 2001; Liu et al., 2003). It is therefore possible that cell-to-cell contact between T-cells and monocytes/osteoclast progenitor cells is also important for osteoclast formation in periodontitis. Interestingly, the expression of RANKL mRNA is up-regulated in gingiva from patients with advanced periodontitis, whereas OPG mRNA is down-regulated (Liu et al., 2003). In situ hybridization showed that RANKL was mainly expressed in lymphocytes and macrophages. This is in agreement with findings demonstrating the lack of RANKL mRNA in fibroblasts isolated from human gingiva (Nagasawa et al., 2002, and our own observations). Interestingly, OPG mRNA is abundantly expressed in human gingival fibroblasts, and the expression is up-regulated by lipopolysaccharide (LPS; Nagasawa et al., 2002). Culture supernatants from LPS-stimulated gingival fibroblasts reduce osteoclast formation in cultured human peripheral blood lymphocytes, indicating that gingival fibroblasts may be involved in a negative control of osteoclast formation in periodontal disease. Further support for the view that RANKL expressed by inflammatory cells is important in periodontitis comes from the observation that stimulation of CD4+ T-cells with Actinomyces actinomycetemcomitans (A.a.), a Gram-negative anaerobic bacterium associated with certain forms of periodontitis, induces RANKL expression (Teng et al., 2000). Transplantation of peripheral blood leukocytes from patients with periodontitis to NOD/SCID mice, followed by oral challenge with A.a., leads to osteoclast formation and local bone resorption in alveolar bone surrounding the teeth. This response can be inhibited by OPG treatment (Teng et al., 2000). Porphyromonas gingivalis, another bacterium implicated in periodontitis, can also induce RANKL expression in lymphocytes, and this is associated with increased osteoclast formation in spleen cell cultures (Jiang et al., 2002). These observations suggest that bacteria can stimulate osteoclast formation directly by activating cell-to-cell interactions in the infiltrating leukocytes, without any involvement of stromal cells/osteoblasts.
In the rheumatoid synovium, fibroblasts express RANKL (Romas et al., 2002b). Similarly, gingival fibroblasts and periodontal ligament cells express RANKL, which can be up-regulated by IL-1 and TNF-α (Sakata et al., 1999), similar to observations in osteoblasts (Hofbauer et al., 1999a). It is therefore possible that not only RANKL-expressing T-cells but also RANKL-expressing resident fibroblasts are important in osteoclastogenesis associated with periodontal disease. Recently, we have observed that RANKL expression in gingival fibroblasts and periodontal ligament cells can be enhanced by the cytolethal distending toxin present in A.a. (Belibasakis et al., submitted for publication). The relative importance of RANKL-expressing T-cells, gingival fibroblasts, periodontal ligament cells, and osteoblasts for inflammation-induced bone resorption is not yet known (Fig. 4), but regardless of cell type, RANK activation is likely to play a crucial role, and therefore inhibition by OPG would be an interesting pharmacological approach to inhibit bone loss in periodontal disease.
Osteoclastic bone resorption is also involved in metastatic lesions in patients with malignant tumors. RANK signaling by RANKL has been suggested to be a crucial factor in cancer-induced osteoclastogenesis. Certain cancer cells express RANKL and might therefore directly induce osteoclast formation, whereas other tumors secrete factors (e.g., parathyroid-hormone-related peptide, macrophage inflammatory protein-1α, IL-6) which can stimulate RANKL expression in osteoblasts (Hofbauer et al., 2001; Kitazawa and Kitazawa, 2002; Oyajobi and Mundy, 2003). Based upon such observations, OPG is now used in clinical trials with the aim to prevent bone resorption in patients with multiple myeloma and breast cancers (Body et al., 2003).
Giant cell granuloma is an uncommon benign skeletal lesion which is also seen in jaw bones. Formation of the giant cells has been suggested to be due to RANKL expression in the stromal cells present in these lesions (Itonaga et al., 2002).
Enhanced numbers of osteoclasts and accelerated bone remodeling are common features for patients with familial expansile osteolysis and expansile skeletal hyperphosphatasia. In both diseases, similar but slightly different base-pair tandem duplications in the TNFRSF11A gene encoding RANK have been found (Hughes et al., 2000; Whyte and Hughes, 2002). The mutations affect the signaling peptide sequence of RANK and are associated with increased RANK signaling, and this may be the reason for the excessive numbers of osteoclasts seen in these patients.
Hyperostosis corticalis deformans juvenilis is a disease with increased and abnormal bone formation secondary to excessive resorption of bone, resulting in debilitating fractures and deformities of the skeleton in infancy or early childhood. Homozygous deletion of the gene on chromosome 8q24.2 encoding OPG (TNFRSF11B) and undetectable serum levels of OPG have been shown in two families with the disease, and are the likely explanation for the uncontrolled differentiation and function of osteoclasts in these patients (Whyte et al., 2002).
Increased osteoclast formation and activation have been implicated in the pathogenesis of post-menopausal osteoporosis, and estrogen deficiency has been shown to be associated with increased osteoclastogenesis (Manolagas and Jilka, 1995). The possibility that estrogen may interfere with RANKL-RANK signaling is therefore intriguing. Direct evidence for this view is the observation that expression of OPG, via estrogen receptor-α, is enhanced in estrogen-stimulated human osteoblasts and the mouse stromal cell line ST-2 (Hofbauer et al., 1999bl Saika et al., 2001). Interestingly, treatment of post-menopausal women with OPG resulted in decreased levels of biochemical markers of bone turnover (Bekker et al., 2001). Estrogen has no effect on RANK expression in the osteoclast progenitor cell line RAW 264.7 (Shevde et al., 2000). However, estrogen has been shown to inhibit osteoclast formation by interfering with signaling pathways downstream of RANK in osteoclast progenitor cells, as shown by decreased MKK-dependent JNK-activity, c-Fos and c-Jun transcription, and c-Fos/c-Jun nuclear translocation (Shevde et al., 2000; Srivastava et al., 2001).
(10) Summary
Osteoclast formation requires proliferation and differentiation of osteoclast progenitor cells, followed by fusion of these mononucleated cells to multinucleated osteoclasts, which subsequently are polarized and activated into mature bone-resorbing osteoclasts. Activation of c-fms, the receptor for M-CSF, and RANK, the receptor for RANKL, in osteoclast progenitor cells is crucial for these processes. Deficiencies of either M-CSF, RANKL, or RANK in mice lead to absence of osteoclasts and an osteopetrotic phenotype. In contrast, deficiency of OPG, a secreted ‘decoy’ receptor blocking RANKL, leads to increased numbers of osteoclasts and an osteoporotic phenotype. The intracellular signaling pathways downstream from RANK in osteoclast progenitor cells involve TRAF6-mediated activation of NF-κB, MAPK, AP-1, c-src, NFATc1, and PI3K/Akt/Bad. RANKL is expressed by stromal cells/osteoblasts and plays an important role in physiological remodeling of bone. The fact that estrogen and glucocorticoids can affect the expression of RANKL-RANK-OPG indicates that uncontrolled regulation of this pathway may be involved in post-menopausal and glucocorticoid-induced osteoporosis. RANKL can also be expressed by T-cells, synovial cells, gingival fibroblasts, periodontal ligament cells, dental follicle cells, and certain cancer cells and has therefore also been implicated in the pathogenesis of bone resorption in rheumatoid arthritis, periodontitis, root resorption, tooth eruption, and skeletal metastasis. The detailed knowledge of the molecular mechanisms involved in RANKL-RANK activation and downstream signaling, as well as the screening for osteoclast-specific genes induced by these pathways, is likely to generate new pharmacological principles for the inhibition of excessive bone resorption in pathological conditions.
Osteoclast progenitor cells are closely related to monocytes/macrophages, and initial proliferation/differentiation is controlled by common signaling pathways. However, several extra- and intracellular signaling molecules are specifically involved in differentiation, fusion, polarization, and activation of the osteoclast-specific lineage. Some of these molecules are expressed in osteoclasts or their progenitor cells (in italic) and some in stromal cells/osteoblasts (underscored). Osteoclasts are derived from hematopoietic stem cells. The osteoclast progenitor cells are closely related to the monocyte/macrophage progenitor cells. Osteoclastogenesis requires initial expansion of the number of progenitor cells induced by activation of c-fms, the receptor for M-CSF. The commitment to the osteoclastic lineage is dependent on activation of the receptor RANK, which is activated by RANKL expressed by stromal cells in bone marrow and osteoblasts in the periosteum. RANK activation results in differentiation of the mononuclear progenitor cells and subsequent fusion to multinucleated latent osteoclasts. Activation of RANK in these cells is required for polarization and activation to mature bone-resorbing osteoclasts. The activation of RANK by RANKL can be inhibited by the RANK-related soluble receptor OPG, released by stromal cells/osteoblasts. Thus, the relative expression of RANKL/OPG is rate-limiting for the osteoclastogenic process. The expression of these molecules is controlled by a variety of hormones and cytokines stimulating bone resorption, e.g., PTH, D3, the IL-6 family of cytokines. Stimulators like PTH and D3 are particularly effective for bone resorption, since activation of their receptors leads to increased RANKL and decreased OPG, whereas members of the IL-6 family of cytokines are less effective, since both RANKL and OPG expression are induced by their receptors. RANKL activates the receptor RANK on osteoclast progenitor cells and mature osteoclasts in a trimeric symmetric complex. In periodontitis, osteoclast formation can be due either to activation of RANKL expressed by infiltrating T-cells or to RANKL induced in resident fibroblasts, periodontal ligament (PDL) cells, or osteoblasts.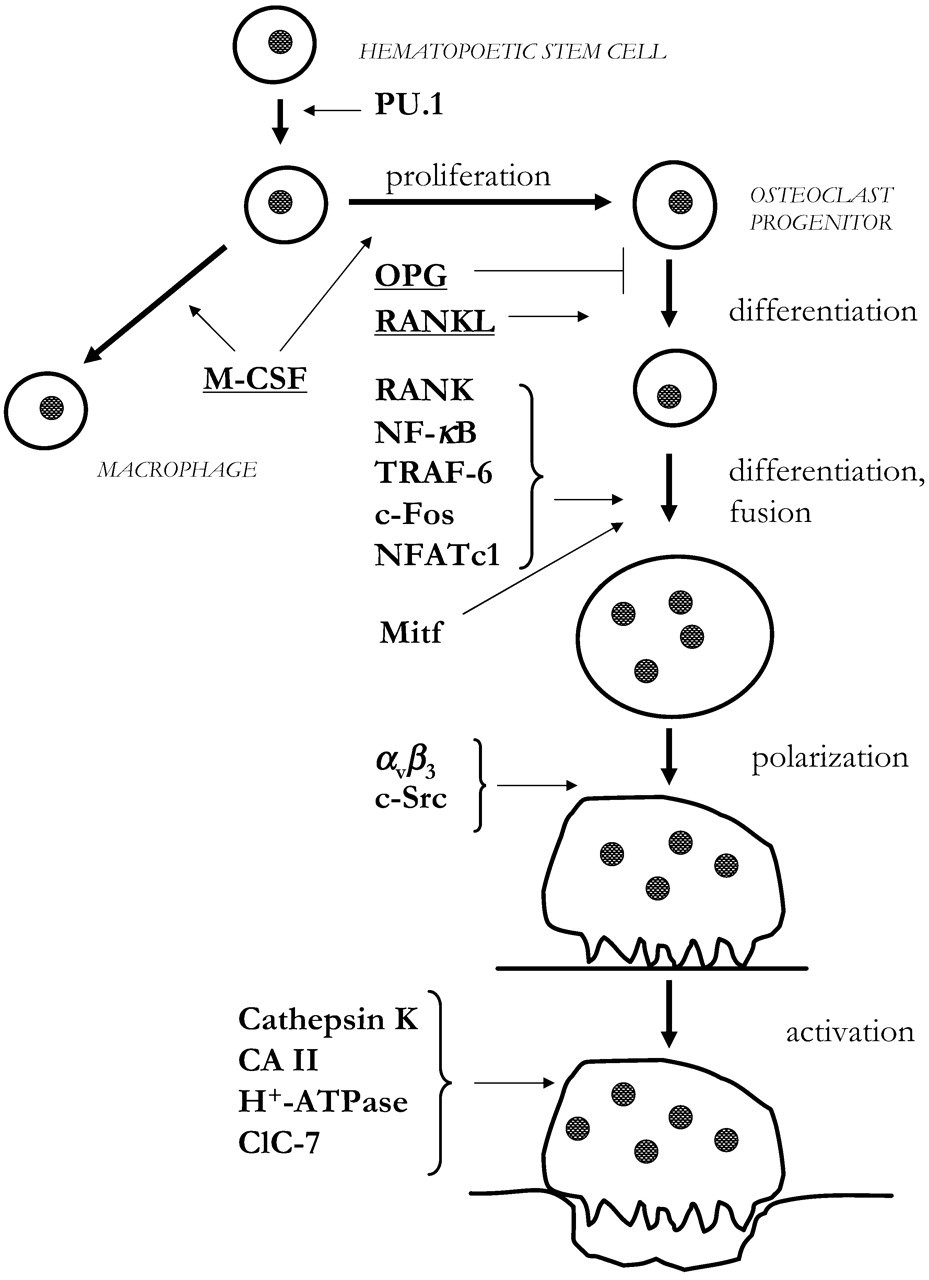
Footnotes
Acknowledgements
Studies performed in the author’s laboratory have been supported in part by The Swedish Medical Research Council, The Swedish Rheumatism Association, The Royal 80 Year Fund of King Gustav V, The County Council of Västerbotten, and the Centre for Musculoskeletal Research, National Institute for Working Life, Umeå, Sweden.