
Abstract
Targeted gene disruption in mice is a powerful tool for generating murine models for human development and disease. While the human genome program has helped to generate numerous candidate genes, few genes have been characterized for their precise in vivo functions. Gene targeting has had an enormous impact on our ability to delineate the functional roles of these genes. Many gene knockout mouse models faithfully mimic the phenotypes of the human diseases. Because some models display an unexpected or no phenotype, controversy has arisen about the value of gene-targeting strategies. We argue in favor of gene-targeting strategies, provided they are used with caution, particularly in interpreting phenotypes in craniofacial and oral biology, where many genes have pleiotropic roles. The potential pitfalls are outweighed by the unique opportunities for developing and testing different therapeutic strategies before they are introduced into the clinic. In the future, we believe that genetically engineered animal models will be indispensable for gaining important insights into the molecular mechanisms underlying development, as well as disease pathogenesis, diagnosis, prevention, and treatment.
Introduction
Significant advances in the study of the human face have revealed the genetic and environmental basis of numerous craniofacial disorders. About seven percent of live births in the United States are affected with some form of congenital disorder. An approximately two to three percent of all infants and children by the age of 5 years are found to have craniofacial birth defects. Of the 6000 known hereditary syndromes, over 700 have either dental or craniofacial defects, and over 250 of them have associated clefting defects (for more details, see http://www.nidcr.nih.gov/about/strat-plan/crani.htm). Because craniofacial development is particularly prone to disturbances and perturbations by teratogenic insults or as a result of genetic mutations, understanding the complicated process of development and fusion of craniofacial tissues has been challenging. However, the last decade has witnessed a rapid progress and expansion in research efforts focused on craniofacial development and disease. This is in part due to new paradigms in related fields such as molecular genetics, developmental biology, and mouse embryology and due to significant advances in recombinant DNA technology, which have provided the necessary tools to answer long-standing questions.
Mouse Models for Understanding Craniofacial Development and Diseases
Complications in the interpretation of genetically engineered mouse models
The laboratory mouse, Mus musculus, is preferred for modern biomedical research because of its small size, short reproductive cycle, known genetic information, and, most importantly, because of our ability to introduce precise genetic alterations into mouse embryonic stem cells and transfer them to stable mouse lines. Therefore, genetically manipulated mice are now widely used as animal models to help us understand how diseases develop and are prevented and treated. Numerous other animal species, such as fly, worm, and zebrafish, have been used for mutagenesis experiments to study developmental and molecular processes; however, mammals such as mice prove to be better animal models, since they are more likely to phenomimic human diseases and disorders. Chromosomal mapping of the murine homologues of candidate genes for many human diseases has been accomplished. However, the identification of such homologues does not guarantee the existence of a spontaneous mutant for a given human genetic disease. The rationale for generating a gene knockout mouse bearing an inactivated or mutated gene is most often based on presumed or unknown functions of the candidate gene from its protein structure, its expression pattern, pharmacological studies, or from the study of a chromosomal locus implicated in the cohorts of patients under study. Following the publication of the first draft of the human genome, many researchers have turned to murine knockout strategies to delineate the in vivo functions of genes of interest and their contributions to disease pathophysiology. Over the past few years, several newly developed knockout mouse models display craniofacial phenotypes. Yet many researchers found unexpected phenotypes. The heady excitement that permeated the early work has given way to a more careful approach. Several criteria have been applied to evaluating potential mouse models, based on phenotypic and genotypic characteristics (Darling and Abbott, 1992). However, any one model often does not reflect all of the phenotypic characteristics of the human disease condition.
Animal models of human disease are important for biomedical research, because they help us understand disease pathogenesis and allow for the development of therapeutic strategies to study the efficacy of novel treatments prior to the conduct of costly and time-consuming human clinical trials. In some cases, the lack of an animal model has hindered progress in the development of effective therapies for debilitating and fatal genetic diseases. For example, our own attempts have yielded benefits in developing novel approaches to treat Fabry disease using a gene knockout model developed in our laboratory (Ohshima et al., 1997; Abe et al., 2000; Takenaka et al., 2000). Additionally, we have also identified novel oral and dental defects in the Fabry mice and Fabry patients (Baccaglini et al., 2001; Dunglas et al., 2001).
Herein, we mainly discuss controversy in the use of genetic engineering technology to understand craniofacial abnormalities. Although gene knockout mouse models provide valuable information about the in vivo gene function, most often concerns have been raised about their intrinsic value as an ’ideal animal model’ for a given disease. Some of these concerns are listed and subsequently discussed below by drawing on lessons from research within and outside the craniofacial field:
Genetic background Early lethality of animals Differences in the life spans of humans and mice Interference by a selection marker gene Compensatory genetic loci Functional redundancy in the in vivo and in vitro conditions
Genetic background
Some of the null mutants have demonstrated a surprisingly high degree of phenotypic variability between individual mouse lines (a few of these mouse models are discussed below). This is probably due to the mixed genetic background from which the mutants are often initially derived. However, several inbred lines are now available for gene knockout studies. While there are other factors to be considered in the making of an ideal knockout mouse model, the properties of the inbred strain in which knockout mice will be generated must be considered before such a long-range project is undertaken (Erickson, 1996; Gerlai, 1996). A mouse model for Branchio-oto-renal (BOR) syndrome is perhaps such an example in the area of craniofacial diseases. Eya-1 heterozygotes show a conductive hearing loss and renal abnormalities similar to that of BOR syndrome, whereas homozygous mice die at birth with reduced head size, open eyelids, and severe craniofacial and skeletal defects accompanied by the absence of thymus, kidney, ear, and parathyroid glands (Xu et al., 1999). However, all newborn Eya-1 null mice in the 129/SvJ and BALB/c backgrounds exhibit secondary cleft palate, whereas, in the C57BL/6J background, the palatal shelves are fused abnormally to the nasal septum. Thus, these results show that in BOR syndrome, in which manifestations vary widely even among family members, the genetic background significantly influences the severity of the associated defects.
A distinctive case of phenotypic variations due to genetic background has also been reported for TGF- β 1 knockout mice. In the mixed 129xCF1 (50:50) background, 50% of the null embryos died before implantation (Kallapur et al., 1999). The other half die of an autoimmune-like multifocal inflammatory disease at weaning (Shull et al., 1992; Kulkarni et al., 1993). Backcrossing the TGF- β 1 -/- alleles into a C57BL/6 background converts partial embryonic lethality to complete lethality, whereas backcrossing to CF1 or C3H does not result in complete embryonic lethality. Only 20% embryonic lethality was observed in TGF- β 1 -/- embryos in a BALB/c background, similar to that seen in the NIH/Ola background (Bonyadi et al., 1997). Moreover, when these animals were maintained in a mixed genetic background of 129xC57BL/6J/Olax129xNIH/Ola, there was partial embryonic lethality occurring during the yolk sac stage (E10.5) but not during the pre-implantation stage (Dickson et al., 1995). TGF- β 2 null mice produced in the Black Swiss strain show severe congenital defects and die perinatally (Sanford et al., 1997). Producing these null mice in an inbred genetic background would probably reduce the number of phenotypes observed. When one colony of TGF- β 3 null mice was kept on a mixed background (129 x CF1; 50:50) and another backcrossed several generations in C57BL/6 strain, the inbred colony displayed more severe clefting as compared with that of the mixed background. The mixed-background colony, with variable expression profiles, provided more information on cleft palate defects (Proetzel et al., 1995). Variable penetrance of the phenotype in a mixed-background colony suggests that there are modifier loci for each phenotype that perhaps can be identified by linkage studies (Doetschman, 1999).
The selection of a strain for making a null mouse may have considerable impact on its phenotype, since unlinked genes in the background strain can significantly affect the disease phenotype. An interesting example was reported by Threadgill et al.(1995) for a null mutation in the epidermal growth factor receptor (EGFR) gene. On the outbred CF-1 background, EGFR mutant embryos show defects in the inner cell mass that cause peri-implantation lethality; on the 129/SvJ inbred background, these mutants have placental defects that cause mid-gestation lethality; and on the congenic C57BL/6J background, defects in various organs cause juvenile lethality. This variability in phenotypes in related genetic backgrounds could potentially help to identify modifier loci that affect the phenotype of interest. Moreover, by further genetic analysis using chromosomal mapping and cloning of unlinked modifier genes, it may be possible to suppress the corresponding diseases. Another example of the effect of genetic background on the usefulness of a null mouse model is the gene knockout mouse models for cystic fibrosis, which show considerable differences in the severity of their phenotypes, ranging from a milder phenotype (Dorin et al., 1992) to a severe phenotype (Snouwaert et al., 1992). These differences are most likely due to leaky expression of the mutated target gene in the mouse model with a milder phenotype. Similarly, retinoblastoma-related p130 null embryos generated in a BALB/cJ genetic background displayed arrested growth and died around E11 to E13. In contrast, null mice on a C57BL/6 background were viable and fertile. It was therefore suggested that p130 in a BALB/cJ genetic background plays an essential role in normal development. Moreover, it is speculated that this could be due to a modifier gene(s), which may have an epistatic relationship with p130 (LeCouter et al., 1998a). Mice lacking p107 developed in a BALB/cJ background were viable and fertile but displayed impaired growth, reaching about 50% of normal weight by 21 days of age. These mutants reverted to a wild-type phenotype following a single backcross with C57BL/6J mice, suggesting the existence of modifier gene(s) that may have an essentially epistatic relationship with p107 (LeCouter et al., 1998b).
Early lethality of animals
Cystic fibrosis results from a defective, or a deficiency in the, cyclic-AMP regulated chloride ion channel, called the cystic fibrosis (CF) trans-membrane conductance regulator (CFTR) (Riordan et al., 1989). In 70% of the patients with CF, the deletion of three bases in exon 10 (F 508) is the major cause of the disease. The most striking feature of CF is attributed to abnormalities of the epithelial surface in the respiratory, digestive, and reproductive tracts. Many patients also show cirrhosis of the liver, accompanied by infertility in males. Death usually occurs as a result of respiratory infection that follows obstruction of the airways by thick, viscous mucous (Boat et al., 1989). A murine model of CF, generated by disruption of the CFTR gene, reproduced many pathological conditions similar to the physiology of the human CF disease. Unfortunately, these animals exhibit more severe symptoms than expected, leading to a shortened life span that is more exaggerated than that observed in humans. It may be possible to observe more symptoms of human CF, such as pulmonary infection, in other related mouse models, months after birth (Snouwaert et al., 1992; Grubb and Boucher, 1999). Similarly, gamma-glutamyl transpeptidase (GGT)-deficient mice display glutathionemia and glutathionuria, as seen in patients with GGT deficiency. However, the severity of disease seen in these mice is much more devastating, leading to early death. A possible explanation could be the presence of multicopy genes for GGT in humans and a single-copy gene in mice. Thus, mutations in one of the human genes may not completely inactivate GGT activity in all organs and may allow for survival with fewer complications (Lieberman et al., 1996).
Differences in the life-spans of humans and mice
Certain human disorders with late onset, e.g., in the second or third decade, may be difficult to model in mice. For example, α-galactosidase-A deficiency causes Fabry disease in humans and results in a progressive deposition of neutral glycosphingolipids that have terminal alpha-linked galactosyl moieties in vascular endothelial cells, causing either renal failure or premature myocardial infarction or strokes. Yet, α-galactosidase-A null mice appear clinically normal, in contrast to Fabry patients, who suffer from an excruciatingly painful and fatal disorder. These mice exhibit typical lipid inclusions with lamellar structures in the lysosomes of target tissues (Ohshima et al., 1997) and also have significant accumulation of neutral glycosphingolipids in the liver and kidney, as is seen in Fabry patients. The disparity in the symptoms seen in human and mice with α-galactosidase-A deficiency poses certain difficulties for in-depth studies; however, the disparity also offers a unique opportunity for the development of preventive or therapeutic approaches to reverse metabolic defects in an otherwise normal animal model.
Interference by a selection marker gene
A targeted disruption of a gene of interest is usually carried out by the introduction of a selection marker at the site of disruption of the coding sequence. The neomycin (neo) resistance gene under the phosphoglycerate kinase 1 (PGK) promoter is the most commonly used selection marker for a null mutation. In some cases, it has been reported that the PGK-neo gene can reduce expression of nearby flanking genes and cause unusual phenotypes (Gingrich and Hen, 2000). Rijli et al.(1994) demonstrated altered expression of the Hoxd-9 gene following disruption of the Hoxd-10 gene by insertion of a PGK-neo gene, illustrating that the targeting of a point mutation into a gene cluster can result in misexpression of a neighboring gene. Targeted mutation in the FGFR3 gene for the generation of a mouse model for achondroplasia also showed interference by the neo resistance cassette with processing of mRNA. These null mice exhibited bone overgrowth, whereas removal of the neo resistance cassette by Cre/loxP recombination produced smaller mice with a shortened craniofacial area and protruding incisors that resembled a human dominant dwarf phenotype (Wang et al., 1999).
Compensatory genetic loci
Some gene knockout mouse models do not show any obvious phenotypes and thereby pose a problem for investigators pursuing any particular approach in their phenotypic analysis. This could occur if the mutated gene is non-functional throughout development. Changes that arise due to the absence of a gene during the entire life span of a mouse could also yield an unexpected phenotype in the mature animal. One possibility could be that the absence of a given gene is compensated for by other gene(s). The ’goosecoid’ gene is implicated in the formation of craniofacial structures. However, goosecoid knockout mice do not display any gross abnormalities except for their lethargy and inability to feed (Yamada et al., 1995). The lack of a gastrulation phenotype suggests potential compensation of the goosecoid gene by another functionally related gene.
The knockout mice for Lef-1, a transcription factor highly expressed in the ectomesenchyme of the tooth bud stage, have shown an early transient arrest of tooth development after formation of the bud and mesenchymal condensation (Kratochwil et al., 1996). However, this does not indicate that Lef-1 has no role at the bud stage, but its role is being possibly compensated for by Tcf-1, a closely related HMG box transcription factor, or some other genes (Oosterwegel et al., 1993).
Prx-1 and Prx-2 (formerly known as Mhox and S8, respectively) are closely related members of the paired class homeobox genes (Opstelten et al., 1991; Cserjesi et al., 1992; Kern et al., 1992; ten Berge et al., 1998). Their expression patterns in mesenchyme are very similar. Prx-1 null animals show striking skeletal defects in the skull, limb, and vertebral column (Martin et al., 1995). However, mice with Prx-2 gene inactivation do not produce any phenotype. Further, Prx-1 and -2 double-mutants show many abnormalities in addition to those observed by Prx-1 null phenotype. Defects were seen in the external, middle, and inner ear, and they also include a loss of skull bones, cleft mandible, and limb abnormalities. Sometimes there is a single or no incisor present in the mandibles, suggesting that Prx-2 is unable to compensate for the Prx-1 gene deficiency (ten Berge et al., 1998).
Targeted mutations in either of two members of the Distal less (Dlx-1 or Dlx-2) homeobox gene family lead to defects in the development of skeletal organs derived from proximal ends of the first and second branchial arches (Qiu et al., 1995, 1997), but do not show any defect in tooth development. However, mice with null mutations in the Dlx-1 and -2 exhibit selective absence of upper molars and an arrest at the lamina stage (Thomas et al., 1997). The expression of Dlx-3, -5, and -6 is restricted more to the distal region. Thus, this explains a possible compensation mechanism for the loss of Dlx-1 or -2, which could not affect mandibular and distal hyoid arches (Depew et al., 1999). This is one of the unique models illustrating a distinct functional redundancy with other Dlx genes expressed in the mandibular but not in the maxillary mesenchyme.
The pleiotropic growth factor FGF-2 is known to be a developmental and homeostatic gene; however, surprisingly, disruption by gene targeting does not produce a distinct phenotype. The FGF-2-deficient mice live a long and healthy life with a normal fertility pattern but with minor vascular abnormalities (Zhou et al., 1998). Interestingly, transgenic mice ubiquitously over-expressing FGF-2 had very short legs, suggesting an important role for FGF-2 in bone development (Coffin et al., 1995), yet the bones in knockout animals are normal. These contradictory results between the two animal models could be due to developmental compensation by other FGF isoforms, which may or may not signal through the same FGF-2 receptor. It should also be noted that loss-of-function and overexpression models are not reciprocal genotypes.
Brown et al.(2000) reported a knock-in strategy involving insertion of inhibin b (inhb b) into the inhibin a (inhb a) locus to rescue the inhibin a null phenotype. Inhb a null mice show failure to develop whiskers, incisors, and mandibular molars, as well as cleft palate, and are subject to neonatal lethality. Inhb b null animals show an eyelid closure defect, prolonged gestation, impaired lactation, normal growth and gonadal differentiation, normal fertility and survival, normal whiskers and tooth development, and no cleft palate. Animals generated by knock-in named Inhb a BK/BK (where a region of inhb a encoding the mature protein was replaced by inhb b) had normal whiskers and tooth development, no cleft palate, symmetrical growth deficiency, enlargement of external genitalia, hypogonadism, sunken eyes, short hair, delayed hair growth, and diminished female fertility. This emphasizes the efficacy of the knock-in strategy to characterize the precise roles of compensatory genes.
Functional redundancy in the in vivo and in vitro conditions
We have selected a few examples from the TGF- β and Hox families highlighting redundancy among family members. In mammalian species, three distinct TGF- β isoforms, -1, -2, and -3, have been well-characterized. The TGF- β superfamily of growth factors regulates diverse biological processes. These isoforms apparently have dual and opposing activities because of their context-dependent nature and their ability to substitute for the other isoform in certain in vitro assays. Under in vivo conditions, they exhibit overlapping, yet distinct, expression patterns in developing tissues and pathological processes (see review by Roberts and Sporn, 1993). They function through the common TGF- β RI/II receptor, which could obviously result in considerable redundancy in their function. However, recently established gene knockouts for each of the three TGF- β isoforms have clearly demonstrated their specific roles during development, with no distinct overlap (Table 1), although redundancy in some tissues cannot be ruled out.
McLain et al.(1992) analyzed the role of murine Hox-2.3 during development using a dominant gain-of-function model. These transgenic mice die shortly after birth and exhibit craniofacial abnormalities, such as open eyes and cleft palate. Ventricular septal defects accompanied by skeletal abnormalities in the bones of the craniocervical transition were also observed. The main deficiencies were observed in the frontal and parietal bones in addition to supra-occipital bones. Interestingly, transgenic mice expressing Hox-1.1 (Kessel et al., 1987; Balling et al., 1989) and Hox-2.2 (Kaur et al., 1992) displayed defects similar to those seen in Hox-2.3 transgenic mice. Thus, Hox-2.3 overexpression causes developmental abnormalities strikingly similar to those found in Hox-1.1 and Hox-2.2 mutants, indicating a high degree of functional overlap between these genes.
BMP-7 is reportedly expressed in dental epithelium during the dental lamina, bud, and cap stages (Åberg et al., 1997). During the bell stage, the mRNA and protein expression profiles of BMP-7 shift from the dental epithelium to the dental mesenchyme. With progressive differentiation of odontoblasts, immunostaining for BMP-7 in the dental papilla becomes more restricted to the layer of fully functional odontoblasts involved in the process of depositing predentin and dentin. In contrast, secretory-stage ameloblasts exhibit weak staining. However, histological analysis of teeth of BMP-7 null mice do not reveal any tooth defects, indicating redundancy in BMP family members (Helder et al., 1998).
Mouse models with phenotypes that conflict with human disease manifestations
Some of the gene knockout mouse models display unexpected phenotypes or no phenotypes, indicating the disadvantages of using such models to duplicate human disease conditions accurately. Although most mutant mice phenomimic human diseases, their direct comparison with human syndromes is sometimes difficult. An appropriate example is the FGFR-3 mutation that generally affects endochondral bones. In humans, three dominant skeletal dysplasias—Thanatophoric dysplasia (TD, mutation in linker region R248C), Achondroplasia (ACH, mutation in transmembrane regions G380R and G375C), and TD II (mutation in tyrosine kinase domain K650E)—result in the most frequent form of dwarfism. These dysplastic disorders display a graded spectrum of phenotypic severity (Bellus et al., 1995; Naski et al., 1996; Webster and Donoghue, 1996; Thompson et al., 1997; d’Avis et al., 1998). In FGFR-3 null mice, there is increased long-bone growth, suggesting that FGFR-3 is a negative regulator of bone growth (Colvin et al., 1996; Deng et al., 1996). Recently, investigators have identified a novel mutation in human FGFR-3 (K650M) which causes severe ACH with developmental delay and acanthosis nigrican (SADDAN). Patients exhibit hyperkeratosis, hyperpigmentation, severe skeletal dysplasia, brain anomalies, and hearing loss (Bellus et al., 1999; Tavormina et al., 1999). Another mutation in adjacent nucleotide A1948G:K650E was reported which causes TD type II, a lethal form of skeletal dysplasia. Iwata et al.(2001) created the corresponding K644M mutation into the mouse FGFR-3 gene to mimic the human K650M mutation. To their surprise, mice heterozygous for the K644M mutation show a phenotype similar to human SADDAN, contradicting an earlier knockout report. This mouse model provided more insights into the molecular mechanism of how mutation in FGFR-3 leads to severe skeletal dysplasia.
Inactivation of the DLX gene in mice also suggests functional redundancy among family members with respect to dental patterning. DLX-1 and -2 individual knockout mice do not exhibit any tooth defects (Qiu et al., 1995), while mice lacking both DLX-1 and DLX-2 exhibit arrested maxillary molar development (Qiu et al., 1997). However, it is not clear whether these two DLX genes really act as selectors or are required for downstream processes in some teeth but not others. Defects of the first branchial arch in DLX-1 and -2 knockout mice are limited to derivatives of the maxillary process, which cannot be categorized as an ideal model for human defects. Similar unpredicted phenotypes were also noticed in goosecoid knockout mice. The expected gastrulation defects were not observed in these mice. Instead, the phenotype was manifested at a late embryonic stage (Yamada et al., 1995). The goosecoid-like (GSCL) gene, which was suspected to cause velocardiofacial syndrome and DiGeorge syndrome (VCFS/DGS) in humans, when knocked out in mice, did not exhibit any phenotypic similarities to VCFS/DGS patients (Wakamiya et al., 1998). Patients with Beckwith-Wiedemann syndrome have mutations in the p57 kip2 gene (Bhuiyan et al., 1999), but analysis of p57 kip2 null mice revealed no similarity to Beckwith-Wiedemann syndrome (Takahashi et al., 2000). Interestingly, knockout mice for individual isoforms of the retinoic acid receptor α1 (RAR-α1) (Li et al., 1993), RAR- β 2 (Mendelsohn et al., 1994), and RAR- γ 2 (Lohnes et al., 1993) did not exhibit any phenotype as anticipated, although RAR- γ mutants exhibit growth deficiency, early lethality, and male sterility, demonstrating high redundancy among the RARs.
Pax-9-deficient mice die shortly after birth and exhibit a wide range of developmental defects (Peters et al., 1998). They lack pharyngeal pouch derivatives and teeth, and they also display craniofacial and limb abnormalities. The secondary palate is clefted, and a variety of skeletal abnormalities develops, affecting the head and the visceral skeleton. The teeth appear as rudimentary buds, with developmental arrest occurring at the bud stage. The severe phenotype of the null mice indicates the complex function of this gene, but a recent report of a human Pax-9 frameshift mutation was associated with oligodontia (Stockton et al., 2000). The affected patients have normal primary dentition but lack most of the permanent molars. This difference in the manifestations of the Pax-9 mutation in mice vs. humans highlights the importance of knowledge concerning the functional mutation of the candidate gene in man. In knockout mice, targeted gene disruption results in a specific functional mutation, whereas in human cases, genetic mutations or re-arrangements do not necessarily yield a complete functional mutation, but perhaps result in a dysfunctional gene product.
Mutations in the Pax-3 gene were identified in patients with Waardenberg syndrome (WS). WS is a dominant pigmentation disorder with congenital deafness. Clinically, WS is classified into type I (WS1, OMIM 193500) and type II (WS2, OMIM 193510), based on the presence of dystopia canthorum in WSI patients (broad nasal root and nasal bridge, hypoplastic alae, increased incidence of spina bifida, and an outward displacement of the inner corner of the eye). Subtype III (WS3, OMIM 148820), or Klein-Waardenberg, was later identified with limb hypoplasia with variant presentation of WS type I. Mutations in Pax-3 and microphthalmia transcription factor (MITF) have been implicated in WS. In mice, Pax-3 is encoded by the splotch (sp) locus (Strachan and Read, 1994; Tassabehji et al., 1994), and the heterozygous (sp/+) mice display white spotting on the abdomen, tail, and feet. Null (sp/sp) mice for splotch alleles die midway through gestation, with defects in the neural tube and spinal ganglia. Hypomorphs may survive to birth and have spina bifida with either abnormal or missing spinal ganglia. Most WS patients with heterozygous Pax-3 mutations have hearing loss, but sp/+ mice have normal hearing capacity with no physiological and morphological defects in the inner ear (Steel and Smith, 1992). The patients homozygous for Pax-3 mutations live at least three months with no neural tube defect (Zlotogora et al., 1995), suggesting adverse effects of gene dosage in the loss-of-function mutations. Interestingly, in humans, Pax-3 gain-of-function mutations cause alveolar rhabdomyosarcoma (Barr et al., 1993; Galili et al., 1993; Shapiro et al., 1993). The other example is Werner syndrome (WRN), an autosomal-recessive disorder characterized by genomic instability and premature onset of several age-related diseases. Interestingly, the murine homologue, mapped to chromosome 8, is in a region syntenic with the human WRN gene, but the homozygous mutant mice appear normal (Lebel and Leder, 1998).
A point mutation in the collagen type X, α1 gene (COL-10 A1), that produces a frame shift mutation causes Schmid metaphyseal chondrodysplasia in humans (Warman et al., 1993). This mutation may prevent the association of the mutant polypeptides during trimer formation, resulting in a decreased amount of normal protein. Mice carrying a dominant-negative mutation in the Col-10 transgene have skeletal deformities with hypertrophic growth plate cartilage and a decrease in newly formed bone. In addition, there is a deficiency of leukocytes in the bone marrow associated with lymphopenia and a reduction in the size of the thymus and spleen. These conditions resemble human chondrodysplasias like spondylometaphyseal dysplasia and metaphyseal chondrodysplasias (Jacenko et al., 1993). However, mice that are homozygous for germ-line null mutations in the Col-10 gene are phenotypically normal (Rosati et al., 1994). These results provide strong genetic evidence that Schmid metaphyseal chondrodysplasia results from dominant-negative mutations in the COL-10 A1 gene and are not due to an overall deficiency in type X collagen. Thus, mice carrying a functional-null mutation in Col-10 do not appear to be appropriate models for human disease. Conversely, transgenic mice that carry dysfunctional mutations in the Col-10 gene may be an ideal model in which the presence of abnormal type 10 collagen is the main cause of abnormal bone growth and development.
In summary, it is important to characterize the mutations underlying human hereditary diseases and disorders. Human genetic mutations include a wide spectrum of defects, such as point mutations with or without causing an amino acid substitution, hypomorphic mutations, and null mutations causing disruption of the coding sequence. If possible, it is best to introduce the identical mutations into the mouse genome to obtain an animal model with the expected phenotype.
Genetic Complexity
Single genes with a pleiotropic role
The most frequent genetic causes of human disease and death are multifactorial. The majority of genes associated with craniofacial defects show multiple phenotypes. Most of them are strongly influenced by multiple genetic and environmental factors. Gene targeting provides opportunities for investigating the effects of predetermined single gene alterations on the pathogenesis of these diseases in a relatively constant genetic background and environment. However, this can sometimes confound the interpretation of knockout phenotypes. For example, holoprosencephaly (HPE) patients exhibit a wide spectrum of phenotypes, such as loss of midline facial structure, mild hypotelorism, cyclopia, a primitive nasal structure, mid-facial clefts, failures of the division of the cerebral hemispheres, and premaxillary agenesis. Mutation in the human Sonic Hedgehog gene (SHH) on chromosome 7 is believed to be the likely cause of HPE (Roessler et al., 1996). Other genes on human chromosomes 2 (ZIC-2), 18 (TGIF), and 21 (SIX-3) are also implicated in HPE (Meisler, 1997; Wallis and Muenke, 2000). Targeted disruption of the SHH gene in mice manifests the phenotypes of HPE in humans (Chiang et al., 1996; Roessler et al., 1996), illustrating identical multiple effects of a single gene disruption occurring in two different species.
In another case, the MSX-1 mutation also exhibits a wide spectrum of phenotypes, such as autosomal-dominant agenesis of the second premolars and third molars (Vastardis et al., 1996) and cleft palate (Lidral et al., 1998), and it is also associated with CATCH-22 syndrome (Thomas et al., 1998). Mutant mice for DNA-binding proteins containing a homeodomain and two zinc finger clusters, such as δ EF-1, exhibit craniofacial abnormalities of neural crest origin, cleft palate, hyperplasia of Meckel’s cartilage, and dysplasia of the nasal septum (Takagi et al., 1998). These phenotypes mimic those in mice mutated for Hox genes such as MSX-1 (Satokata and Maas, 1994), Mhox (Martin et al., 1995), and Hoxa-2 (Gendron-Maguire et al., 1993). The commonalities in these mutant phenotypes and the expression patterns of δ EF-1 and Hox genes implicate possible interactions between their genetic loci or between their protein products. Such a pleiotropic role has also been noticed in models for the Pitx-2, Prx-1 genes, etc. Deletion of the Pitx-2 homeobox gene by homologous recombination causes abnormal mandibular facial prominence, unusual maxillary development, and cleft palate. Pitx-2 null mice also exhibit mandibular and maxillary tooth arrest similar to that seen in Rieger syndrome (Lin et al., 1999; Lu et al., 1999). Deletion of peroxisomal matrix protein receptor (Pxr-1), a protein that binds peroxisome targeting signal 1 (PTS-1)-carrying proteins, also shows multiple phenotypes in mutant mice and is a suitable model for Zellweger disease (Baes et al., 1997). Therefore, one needs to take extra precautions in the interpretation of the observed phenotype in genetically altered mice or other lower species. This could be misleading when matched with the human disease condition.
Multiple genes associated with a common syndrome
Multifactorial disorders imply the involvement of multiple genetic loci interacting with environmental factors that result in complex phenotypes. In other words, multiple genes are likely to be involved in congenital birth defects. This may be due to disruption of signal transduction pathways at multiple steps that are required for the formation of certain anatomical structures. Therefore, when the effects of gene disruption are primarily restricted to a specific tissue, it is relatively simple to search for a human disease state that resembles the animal model. However, when genes exhibit a high level of pleiotropy, then the phenotype can become very complex for deciphering the precise etiology. Examples of genes with pleotropic roles discussed above are listed in Table 2.
Comparative gene mapping
When this review was written, more than 31,581 genes in the mouse had been identified, and about 17,357 genes had been mapped. Yet only 7392 gene homologies of mouse/human have been studied so far (ftp://ftp.informatics.jax.org/pub/informatics/reports/MGD_Stats.sql.rpt). If a mouse mutation has been mapped, the map position of the human analogue can most likely be predicted. Therefore, once the map location of the mouse mutant is known, comparative mapping information can be used to predict the likely location of the candidate gene for a human genetic disorder (Darling and Abbott, 1992; DeBry and Seldin, 1996; Bedell et al., 1997a,b).
Information obtained from mouse models has led directly to the identification of human disease genes for Waardenburg syndrome. The MITF mutation has been reported in WS patients. The Mitf gene is encoded by the mouse microphthalmia (mi) locus (Jackson, 1994; Moore, 1995). All ’mi’ alleles cause defects in melanocytes, which lead to defective pigmentation and deafness. A mutation identical to that of the original mutation at this locus, the ’mi’ allele, was found in a family with WS-2 (Tassabehji et al., 1994).
Some syndromes have high penetrance but variable manifestations. In these situations, a precise phenotypic description can assist investigators in predicting the genotype. In such conditions, affected individuals have different phenotypes, yet share the same abnormal gene, or they may look quite similar but have mutations on different genes. For example, in Waardenburg type 1 syndrome, there is uneven spacing of the eyes, deafness, and uneven skin coloration. Waardenburg type 3 has craniofacial-hand-deafness abnormalities. However, both conditions are associated with mutations in the same gene, PAX-3, a paired-box and homeobox gene. PAX-3 is located on chromosome 2q35. Similarly, Waardenburg type 2 has the same features as types 1 and 3, except that the eye placement is normal and the mutation is in another gene, i.e., the MITF gene. Therefore, a mouse that has been suggested as a possible model for Waardenburg type 2 is the MITF mutant (Asher and Friedman, 1990).
Four different types of Waardenburg syndrome (WS) have been identified based on phenotypes. One or more mouse mutant alleles, e.g., Ph (patch), s (piebald), Sp (splotch), m (microphthalmia, mitf), were reported homologous to mutations causing WS in humans (Asher and Friedman, 1990). WS-1 maps to human chromosome 2q37 (Foy et al., 1990), and Sp maps to mouse chromosome 1. Although phenotypically they do not appear to match, chromosomal mapping of WS-1 and Sp indicated their close relationship. Interestingly, Sp has also been reported to be caused by mutation in the murine Pax-3 gene, and indeed some families with WS-1 have mutations in the human homologue of the Pax-3 gene (Baldwin et al., 1992; Tassabehji et al., 1992).
In some instances, localization of the human disease gene is used to predict a gene responsible for a mutant mouse phenotype. One of the cited examples is from X-linked agammaglobulinemia, Bruton type in humans (Tsukada et al., 1993; Vetrie et al., 1993), and X-linked immune deficiency (xid) in mice (Rawlings et al., 1993). Human DSPP (located on chromosome 4q21.23-q21.23) mutations are known to cause dentinogenesis imperfecta (Xiao et al., 2001; Zhang et al., 2001), and a mouse dspp (chromosome 5) mutation developed in our laboratory results in a similar condition (Sreenath et al., 2003a). Ectodermal dysplasia (EDA) in humans and Tabby (Ta) in mice are both X-linked dominant mutations. EDA patients show hypotrichosis, sparse scalp hair, absent sweat glands, conical teeth, and respiratory infections. This gene is mapped to Xq12 in humans (McKusick, 1990) and shows homology to the ’Ta’ mutation in the mouse X chromosome. ’Ta’ mice show phenotypes of hair and teeth defects similar to those in humans, including respiratory disorder.
The mouse mutants Hyp and Gyro (Gy) are known to be ideal models for hypophosphatemic Vit D-resistant rickets type 1 (HPDR-I), based on the phenotype and mapping of human Xp21.1-p22.3 and the mouse X chromosome (Eicher et al., 1976; Lyon et al., 1986). Greig syndrome is an autosomal-dominantly inherited human disease with digital malformation and polysyndactyly. This gene was mapped to chromosome 7p13-p12.3 near the T-cell receptor γ gene (TCRG) and has now been shown to encode a zinc finger protein, GLI-3 (Vortkamp et al., 1991). A similar dominant mutation is observed in the Xt (extra toes) mouse, which maps to chromosome 13, in a conserved syntenic group with human chromosome 7p and is closely linked to TCRG (Kang et al., 1997). Cleidocranial dysplasia (CCD) is an autosomal-dominant disorder in humans characterized by defective bone formation, including dental defects such as supernumerary teeth and delayed eruption of permanent dentition, and is caused by a mutation in the core-binding factor activity (Cbfa-1) gene. Cbfa-1 homozygous null mice show misshapened and severely hypoplastic tooth organs (Mundlos et al., 1997; Lee et al., 1997; D’Souza et al., 1999; Shapiro, 1999). A recent study, however, indicated that the transcription factor Osf-2/Cbfa-1 serves as a master gene regulating osteoblast-specific gene expression. The human and mouse mutant syndromes map to syntenic regions on chromosomes 6p21 and 17, respectively (Mundlos et al., 1995; Otto et al., 1997).
Although, Greig, Xt, and CCD syndromes as well as numerous other mouse mutants were proposed as models based on phenotypic similarities with human diseases, the results of comparative mapping and molecular analysis validated the usefulness of each model. Comparative mapping can lead to a more precise way of studying disease homologies between mice and humans, thereby opening new avenues to better therapeutic potentials.
Reflections on Animal Modeling Approaches for Craniofacial Disorders
Almost all classes of human disorders have been replicated in mice based on either spontaneous or induced mutations or by means of experimental manipulations involving either induced pathogenesis, surgery, physiological systems, or environmental changes. These animal models represent hereditary diseases, diseases due to infectious agents, sporadic cancers, autoimmune disorders, and neurodegenerative disorders. The genetically engineered models for craniofacial diseases and disorders have been generated either by insertional mutations using a pathogen or by genetic modifications in ES cells or fertilized eggs (Table 3):
genes associated with craniosynostosis models for branchial arch and cranial neural crest diseases animal models for dental diseases models for secondary cleft palate
Genes associated with craniosynostosis
Craniosynostosis syndrome is the most common developmental disorder that causes an abnormal shape of the skull due to the premature fusion of one or more cranial sutures (Muller et al., 1997; Wilkie, 1997). Its prevalence is from 1/2100 to 1/2500 births (Hunter and Rudd, 1976; Lajeunie et al., 1995). There are numerous genes that have been implicated in syndromic and non-syndromic craniosynostosis. These genes include the muscle segment of homeobox gene-2 (MSX-2), fibroblast growth factor receptor (FGFR)-1, FGFR-2, FGFR-3, ALX-4, and TWIST (Table 4).
The Boston-type craniosynostosis is caused by dysregulation of the MSX-2 gene product. Expression of a mutated MSX-2 gene (Pro148 → His mutation) in transgenic mice is exhibited by precocious fusion of cranial bones (YH Liu et al., 1995). Overexpression of the wild-type MSX-2 gene has also produced phenotypes similar to that of Boston-type craniosynostosis in mice, consistent with the possibility that the mutation enhances the normal activity of MSX-2 (Ma et al., 1996) and provides a useful model for understanding the pathogenesis of this disease. MSX-2 null mice exhibit prominent phenotypic features reminiscent of abnormalities associated with human MSX-2 haploinsufficiency in the parietal foramina (PFM) (Satokata et al., 2000). The fact that PFM results from MSX-2 haploinsufficiency in man but not in mice suggests different species-specific requirements for MSX-2 dosages. The phenotype of MSX-1/MSX-2 double-knockout mice indicates that MSX-2 function is redundant with that of MSX-1 in calvarial morphogenesis and in the formation of mammary gland, tooth, and hair follicle (Maas and Bei, 1997).
Similarly, point mutations in fibroblast growth factor receptor-1 (FGFR-1) and FGFR-2 have been associated with Pfeiffer (Muenke et al., 1994), Jackson-Weiss (Jabs et al., 1994), and Crouzon syndromes (Reardon et al., 1994), the autosomal-dominant craniosynostosis syndromes that also exhibit variable degrees of limb abnormalities (Muenke and Schell, 1995). Some of the diseases are also believed to be the result of gain-of-function genes or chromosomes, e.g., trisomy. Therefore, the FGFR-1 transgenic mouse that recapitulates the human phenotype may be a more useful model for the study of human diseases. In contrast, mice heterozygous for the FGFR-1 null mutation are normal, and homozygous null mice display early embryonic mortality (Deng et al., 1994; Yamaguchi et al., 1994) and consequently do not appear to be suitable models for these syndromes. Deletion of the TWIST gene causes features similar to those of Saethre-Chotzen syndrome, one of the most common autosomal-dominant forms of craniosynostosis (el Ghouzzi et al., 1997; Bourgeois et al., 1998). Interestingly, this syndrome can also be caused by mutations in FGFR-2 and FGFR-3 (Muenke et al., 1997; Paznekas et al., 1998). Recently, six new mutations of the tyrosine kinase region and a single mutation of the Ig II domain of FGFR-2 that cause craniosynostosis have been identified in humans, in addition to the known mutation hotspot in the IgIIIa/IIIc region (Kan et al., 2002). Studies of the morphogenetic signaling pathways suggest a functional link between TWIST and members of the FGFR family in humans (Paznekas et al., 1998). All of these mouse models for craniosynostosis provide additional opportunities for understanding the link among the signaling pathways of MSX-2, FGFRs, and TWIST involved in suture maintenance and fusion.
Models for branchial arch and cranial neural crest diseases
Development of the branchial arch and cranial neural crest has been studied extensively in craniofacial biology because of the relatively large numbers of human congenital syndromes attributed to branchial and cranial neural crest defects. For example, Waardenburg’s syndrome (Waardenburg, 1951), Treacher-Collins syndrome (Sulik et al., 1987), and Pierre-Robin syndrome (Dennison, 1965) are characterized by abnormal facial features and are thought to involve defects of the first and second branchial arches. The phenotype of endothelin-1 (ET-1) null mice is quite similar to the human congenital diseases known as first pharyngeal-arch syndromes, including Pierre-Robin syndrome and Treacher-Collins syndrome. The syndromes are characterized by morphological abnormality of the tissues derived from the first pharyngeal arch, such as the mandible, ear, palate, and eye, and are thought to be due to an abnormality in the development of specific neural crest cell lineages. ET-1 null mice therefore may be a useful tool for the investigation of the development of the branchial arch and to clarify the etiology of the first branchial arch syndromes (Kurihara et al., 1994). Deletions of ET-1, dHAND, eHAND, and MSX-1 show overlapping phenotypes of velocardiofacial syndrome (VCFS, OMIM 192430), DiGeorge syndrome (DGS, OMIM 188400), and conotruncal anomaly face syndrome (CAF, OMIM 217095), collectively known as CATCH-22 syndrome (Wilson et al., 1993). VCFS and DGS syndromes are reported to be caused by the deletion of one allele on chromosome 22q11 (Driscoll et al., 1993; Saint-Jore et al., 1998) in more than 80% of the affected individuals. Deletion of these genes in mice reveals that development of the neural-crest-derived branchial arch ectomesenchyme is mediated by sequential molecular pathways of signaling peptides (Thomas et al., 1998). Epithelial secretion of ET-1, which enhances dHAND and eHAND gene expression, in turn regulates MSX-1 in the distal branchial arch. Partial disruption of this molecular pathway results in CATCH-22 syndrome. However, deletion of the goosecoid-like (GSCL) gene, which is also reported to cause VCFS/DGS in humans, does not exhibit any of the anatomical abnormalities in homozygous null mice as seen in VCFS/DGS patients (Saint-Jore et al., 1998; Wakamiya et al., 1998), and hence homozygous null mice may not prove to be an ideal animal model. However, it appears that hemizygosity of GSCL, in combination with hemizygosity for other genes in 22q11, contributes to some of the developmental abnormalities seen in VDGF/DG patients.
Animal models for dental diseases
About 30 genes are known to be involved in dental anomalies (Slavkin, 1995). However, very few animal models are available that mimic them. Sharpe (1995) has proposed an explicit, combinatorial model for studying patterning of the mandibular dentition, in which overlapping expression domains of MSX-1, MSX-2, DLX-1, and goosecoid (gsc) specify the location of the dental lamina and the tooth type within it (also see Dolle et al., 1992; MacKenzie et al., 1992; Gaunt et al., 1993). For example, MSX-1, but not MSX-2, is expressed in the dental lamina, whereas molar fields express DLX-1 but not gsc, and incisors express gsc but not DLX-1. However, no homeotic transformations or tooth defects of any kind are reported in gsc knockout mice (Yamada et al., 1995).
A missense mutation (Arg31Pro) of MSX-1, a human homeobox gene, is reported to cause an autosomal-dominant agenesis of second premolars and third molars in humans (Vastardis et al., 1996). However, null mice lacking this gene exhibit multiple craniofacial anomalies, including complete tooth agenesis, cleft secondary palate, and nasal and middle ear abnormalities (Satokata and Maas, 1994). Mice deficient for Msx-2 exhibit late defects in cuspal morphologies, root formation, and enamel organ differentiation (Satokata et al., 2000). Interestingly, MSX-1/MSX-2 double-mutants display more severe phenotypes and exhibit earlier tooth defects than either mutation separately (Maas and Bei, 1997). Knockout models for other genes such as Dlx-1, Dlx-2, and Dlx-5 also exhibit either malformation or arrest of the lamina stage in maxillary tooth development (Bei and Maas, 1998; Acampora et al., 1999; Depew et al., 1999). These models, however, are consistent with a role in epithelial-mesenchymal interaction in early tooth development but not in determining the location or type of teeth. It is also speculated from studies of these knockout models that a midline signaling system, in which SHH and Pax genes are involved, is implicated in patterning the mesio-lateral axis of at least the premaxillary dentition (Chiang et al., 1996). However, one of the problems in studying tooth development in mice is that mice have a highly modified dentition relative to that of other mammals (Butler, 1993). It may be more appropriate to study the expression patterns of candidate regulatory genes in other mammalian species with less modified dentitions.
Three mouse models generated in our laboratory mimic most common dental disorders, such as dentin dysplasia, dentinogenesis imperfecta, and amelogenesis imperfecta. These models are described in detail in the following pages.
Transforming growth factor-β1 (TGF- β 1), a multifunctional growth factor, is expressed in a wide variety of developing tissues from the early embryonic stages. The role of TGF- β 1 in the regulation of cell proliferation, differentiation, embryonic development, and apoptosis is well-characterized (McCartney-Francis et al., 1998; Piek et al., 1999; Massagué and Chen, 2000). During mouse tooth development, TGF- β 1 is expressed initially in the oral epithelium at embryonic day 13 (E13); later, its expression extends into the mesenchymal compartment and then becomes restricted to the ectomesenchymally derived odontoblasts. The odontoblast-restricted expression of TGF- β 1 continues throughout life in mice (Vaahtokari et al., 1991). Odontoblasts produce dentin extracellular matrix (DECM) from E16 that subsequently mineralizes in an orderly manner. TGF- β 1 has been shown earlier to have mitogenic effects in tooth explant cultures (Sloan and Smith, 1999) and to induce secretion of DECM components. Although it has been suggested that TGF- β 1 plays a crucial role in dental tissue repair processes by the induction of reactionary (Smith et al., 1995) and reparative (Tziafas and Papadimitriou, 1998) dentinogenesis, the precise in vivo functions associated with its continued expression are not clearly understood. We and others previously generated TGF- β 1 knockout mice (Shull et al., 1992; Kulkarni et al., 1993), which proved unsuitable for tooth studies because of the maternal transfer of TGF- β 1 through the placenta and milk (Letterio et al., 1994) and because of their perinatal lethality due to massive multifocal inflammation (for review, see Kulkarni et al., 2002). To delineate the in vivo role of this growth factor in tooth development, we generated transgenic mice with the targeted overexpression of active TGF- β 1 to odontoblasts, starting from E17, using the upstream regulatory sequences of the dentin sialophosphoprotein (dspp) gene (Sreenath et al., 1999; Thyagarajan et al., 2001). These mice develop a novel phenotype that resembles hereditary dental disorders, such as dentinogenesis imperfecta-II (DGI) and dentin dysplasia (DD) (Fig.). The dTGF- β 1 transgenic mouse phenotype is associated with significant undermineralization of teeth, dysregulation of dental ECM proteins, and dramatic loss of expression of the dentin sialophosphoprotein gene. Interestingly, these studies also reveal novel expression of crystallins in mouse teeth and their regulation by TGF- β 1 (Thyagarajan and Kulkarni, 2002).
X-linked amelogenesis imperfecta (AI) is an inherited enamel disorder characterized by hypoplastic defects (thin, pitted, or grooved enamel) and/or hypomineralization, and a decrease in enamel mineral content. Several mutations in the human X-linked amelogenin gene have been associated with AI (Lagerström et al., 1991; Aldred et al., 1992; Lench et al., 1994; Lagerström-Fermer et al., 1995; Lench and Winter, 1995; Collier et al., 1997; Ravassipour et al., 2000). X-linked AI provides strong evidence that amelogenin is critical for normal enamel formation. We have recently reported targeted disruption of the amelogenin locus in the mouse genome (Gibson et al., 2001). Amelogenin null mice are normal at birth, but, as early as two weeks of age, they develop a chalky-white discoloration of teeth followed by enamel hypoplasia and cuspal attrition in the molars. The hypoplastic enamel in these mice contains an elemental composition consistent with hydroxyapatite-like minerals, indicating that amelogenins are apparently not required for the initiation of crystal formation. However, the characteristic ’prism’ pattern is completely absent, suggesting a critical role of amelogenins in enamel organization. A review article by Paine et al.(2001) is very informative toward improving our understanding of the advances in genetic, molecular, and structural aspects of enamel biology in the normal mouse.
Dentin sialophosphoprotein (DSPP) is mainly expressed in pre-ameloblasts and odontoblasts of developing teeth, and it is believed to be a candidate gene involved in causing dentinogenesis imperfecta (Xiao et al., 2001; Zhang et al., 2001). Our current studies involving DSPP null mice indicate that DSPP is likely to have a much broader involvement in the formation and mineralization of dentin (Sreenath et al., 2003a). With the availability of tooth-specific DSPP-Cre mice, we anticipate rapid progress in the characterization of the functions of many important genes involved in tooth development and diseases (Sreenath et al., 2003b).
Models for secondary cleft palate
The secondary palate in mammals forms through a complex process characterized by a series of events involving cell proliferation and migration, cell differentiation, production of extracellular matrix, and cell death (Ferguson, 1988). Recently, studies with targeted mutations in mice have revealed a growing number of genes that play crucial roles in this process. Clefts of the primary and secondary palates are the most frequent forms of head and neck malformations in humans, affecting approximately 1-2/1000 live births (Hagberg et al., 1998; Tolarova and Cervenka, 1998). There are about 20 genes involved in clefting defects (Slavkin, 1995). Earlier studies have demonstrated the association of cleft palate with TGF- α , MSX-1, and TGF- β 3 (Shiang et al., 1993; Proetzel et al., 1995; Lidral et al., 1997; Koo et al., 2001). The cleft palate has also been observed in many mouse models with mutations of homeobox genes, like Hoxa-2 (Rijli et al., 1993; Nazarali et al., 2000), Mhox (Martin et al., 1995), Dlx-1 and Dlx-2 (Qiu et al., 1997), and the Lhx-8 gene (Kitanaka et al., 1998). Lhx-8, a LIM homeobox gene, has been mapped to mouse chromosome 3 (Kitanaka et al., 1998); homologues of genes in that region have been mapped to human chromosome 4q25-31, a region that has been linked to craniofacial clefting (Mitchell et al., 1995). The cleft palate phenotype observed in Lhx-8 mutant mice suggests that Lhx-8 is a candidate gene for the isolated non-syndromic form of cleft palate in humans (Zhao et al., 1999). Similarly, mouse mutants for glutamic acid decarboxylase-1 (Gad-1) or for the beta-3 subunit of the gamma-aminobutyric acid A receptor (Gabrb-3) also show a cleft palate phenotype in mice. This suggests that genetic and/or environmental perturbation of GABA pathways during fetal development can alter palatogenesis. Therefore, genes encoding GABA receptors are believed to be likely candidates for human cleft palate (Culiat et al., 1995; Scapoli et al., 2002). Targeted deletion of the twirler (Tw) gene also results in defects of the midfacial region, i.e., clefts of lip, palate, and a disrupted nasal cavity. This twirler mouse model will be valuable to further our understanding of the genetic mechanisms involved in normal development of the mid-facial region (Gong et al., 2000; Gong and Eulenberg, 2001). Table 5 summarizes a list of genes identified in clefting syndromes.
Conclusions and Future Prospects
Gene targeting is a powerful tool for the engineering of murine models for human diseases, and it will continue to have an enormous impact on our understanding of craniofacial development and diseases. Not only do these murine models in many instances faithfully recapitulate the phenotype of the human disease, but they also allow us to study the disease on a uniform genetic background. Some of the models that do not reproduce the exact phenotypes of human diseases are still of considerable interest and value, because of their usefulness in furthering our understanding of important interspecies differences. Since more questions than answers are emerging, the complexity of the mammalian genome is becoming clearer. However, the gene knockout approach must be used with caution, particularly in interpretations of the phenotypes that are obtained in craniofacial and oral biology, where many genes have pleiotropic roles. Comparative mapping could provide valuable information to match mouse and human disorders accurately, and these data would pave the path to testing and developing therapies toward treating human diseases. Mouse models also offer an important approach for testing different treatment strategies before they are introduced into the clinic. In the near future, we are likely to see successful applications of genetic engineering to alter other mammalian genomes to generate animal models in higher species. In this century, we believe, genetically engineered animal models will play an increasingly important role in providing insights into the molecular mechanisms underlying many diseases and also for developing novel therapeutic approaches to prevent and treat many debilitating disorders and diseases.
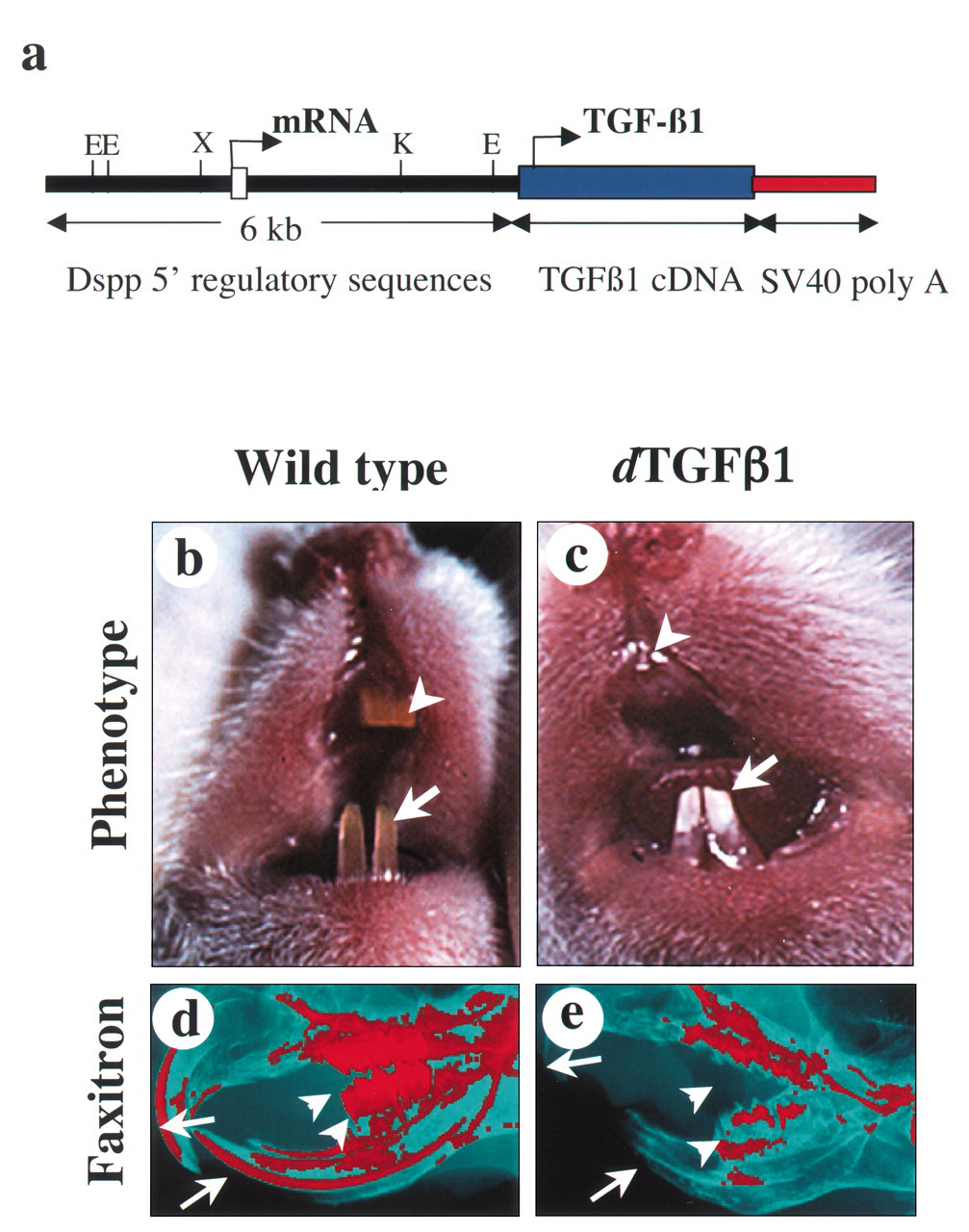
Strategy for generation of dTGF-
β
1 mice and gross tooth abnormalities in these mice.
Footnotes
*
current address, Reliance Life Sciences, Mumbai, India;
Acknowledgements
We thank Drs. Lillian Shum, Taduru Sreenath, Ken Yamada, Yoshi Yamada, and Marian Young for their valuable suggestions and comments during the preparation of this review, and the Journal of Biological Chemistry for permission to reproduce the Fig.