
Abstract
Members of the transforming growth factor-β (TGF-β) superfamily regulate cell proliferation, differentiation, and apoptosis, and control the development and maintenance of most tissues. TGF-β signal is transmitted through the phosphorylation of Smad proteins by TGF-β receptor serine/threonine kinase. During craniofacial development, TGF-β may regulate the fate specification of cranial neural crest cells. These cells are multipotent progenitors and capable of producing diverse cell types upon differentiation. Here we summarize evidence that TGF-β ligands and their signaling intermediates have significant roles in patterning and specification of cranial neural crest cells. The biological function of TGF-β is carried out through the regulation of transcriptional factors during embryogenesis.
Introduction
Embryonic development results from an ordered series of gene interactions, each in turn designating individual cell-type proliferation and differentiation. Central to the regulation of embryonic development is the determination of how signaling and reception of time- and position-restricted instructions occur during morphogenesis of different phenotypes. It is now evident that a hierarchy of growth factors and their downstream transcription factors regulates the expression of genes responsible for determining cell phenotypes during embryogenesis (Edelman, 1985; Lumsden and Krumlauf, 1996). Critical components of the hierarchy have been identified in recent studies on the regulation of craniofacial morphogenesis. Transforming growth factor-β (TGF-β) ligands are expressed in a time- and tissue-specific manner and are important in regulating the formation of various craniofacial structures (Chai et al., 1994; Kaartinen et al., 1995; Proetzel et al., 1995; Sanford et al., 1997). Smad proteins relay TGF-β signaling from cell membrane to nucleus. Extracellular TGF-β signals are transduced via membrane-bound TGF-β type II and type I receptors, which phosphorylate intracellular Smad2 and Smad3. Smad2, 3, 4, and 7 have been associated with the TGF-β signaling pathway. Activated Smad molecules appear to regulate the expression of transcription factors and affect the transcriptional status of target genes (for review, see Massagué 1998, 2000).
Cranial neural crest (CNC) cells migrate ventro-laterally as they populate the branchial arches during craniofacial development. The proliferative activity of these CNC cells produces the discrete swellings that demarcate each branchial arch. Like many developmental processes, the development of facial structures is dependent upon the successful contribution of CNC cells. Although both intrinsic and extrinsic regulatory signals are critical for the proper migration and proliferation of this CNC cell population, recent studies have suggested that the fate specification of CNC cells might be instructed by surrounding tissues during or after migration as these group of progenitor cells contribute to the formation of various craniofacial structures (Couly et al., 2002; Trainor et al., 2002). The post-migratory CNC cells may differentiate into an array of phenotypes following the proper epithelial-mesenchymal interaction and contribute to the formation of various head and neck structures. Various growth (such as TGF-β) and transcription factors (such as Msx1 and 2) have been implicated during the specification and fate determination of neural crest cells. Here we review the biological function of TGF-β signaling molecules and transcription factor Msx1 and 2 in regulating the fate of post-migratory CNC and explore the possible functional significance of the hierarchy of these regulatory molecules during craniofacial morphogenesis.
(I) The TGF-β Signaling
TGF-β is an important regulator during early craniofacial development
Members of the transforming growth factor-β superfamily mediate a wide range of biological activities, including cell proliferation, differentiation, extracellular matrix formation, and induction of homeobox genes, suggesting that TGF-β signaling is important in pattern formation during embryogenesis. TGF-β subtype expression is conspicuous in tissues derived from cranial neural crest cells during early mouse craniofacial development (Heine et al., 1987; Massagué, 1990). In particular, the presence of TGF-β subtypes is obvious in mesenchyme during critical epithelial-mesenchymal interactions related to the formation of tooth organ and Meckel’s cartilage during mandibular morphogenesis (Hall, 1992; Chai et al., 1994). Although the different TGF-β subtypes are indistinguishable in the majority of biological assays with cell lines cultured in vitro, studies have shown that, in a more complex system involving epithelial-mesenchymal interactions, the TGF-β subtypes differ in their biological activities (Chai et al., 1994). This conclusion is further supported by the non-overlapping phenotypes associated with the null mutation of each one of the TGF-β isoforms. In particular, TGF-β1 null mutation results in defective hematopoiesis, vasculogenesis, and pre-natal lethality, indicating that TGF-β1 plays a pivotal role in regulating progenitor cell differentiation and cellular modulation during early embryogenesis (Dickson et al., 1995). During tooth development and dental pulp repair, TGF-β1 is essential for maintaining the homeostasis of the dentin-pulp complex (D’Souza et al., 1998). Targeted disruption of the TGF-β2 gene results in a wide range of developmental malformations, including cardiac, lung, craniofacial, limb, spinal column, eye, inner ear, and urogenital defects. Interestingly, many affected tissues have neural-crest-derived components and simulate neural crest deficiencies (Sanford et al., 1997). Mice carrying the TGF-β3 null mutation have abnormal lung development and cleft palate, indicating defects of epithelial-mesenchymal interactions (Kaartinen et al., 1995; Proetzel et al., 1995). Overall, the TGF-β knockout phenotypes are quite distinct, suggesting that certain regulatory functions mediated by a particular TGF-β isoform cannot be compensated for by other members of the family.
TGF-β IR and TGF-β IIR in TGF-β signaling
Both TGF-β IR and IIR (type I and type II receptor, respectively) are members of a transmembrane serine/threonine kinase family. The third member of the TGF-β receptor family, type III (betaglycan), has a high binding affinity for TGF-β isoforms and may be critical for keeping TGF-β at the cell surface for the type I and II receptors, but has no intracellular signaling motifs (Massagué, 1992; Lin and Lodish, 1993; Lopez-Casillas et al., 1993). The TGF-β ligand binds to the TGF-β type II receptor and triggers heterodimerization with a TGF-β type I receptor (Massagué, 1996). Following heterodimerization, the TGF-β IIR serine/threonine kinase transphosphorylates the type I receptor, resulting in the propagation of a phosphorylation signal to downstream substrates Smad2 and Smad3 (Fig. 1) (Wrana et al., 1994; Derynck and Zhang, 1996). The evidence to date argues that the type II receptors are unable to generate responses independently of type I receptors. It has been proposed that different TGF-β responses may require different levels of signaling and, thus, are inhibited to various degrees by a dominant-negative receptor (Feng et al., 1995; Massagué, 1996). Nevertheless, overwhelming evidence supports the notion that both TGF-β IR and TGF-β IIR are indispensable in eliciting TGF-β biological responses (Wrana et al., 1992; Attisano et al., 1993).
The critical role of TGF-β receptor during TGF-β signaling has been demonstrated in several recent studies. The kinase subdomain of TGF-β type I receptor determines the TGF-β intracellular signaling specificity (Feng and Derynck, 1997). Antisera directed against a TGF-β IIR N-terminal peptide that perturbs TGF-β ligand-receptor binding increases lung branching by 70%. Addition of exogenous TGF-β1 is unable to overcome the stimulatory effects of either TGF-β IIR immunoperturbation or antisense oligodeoxynucleotide treatment on lung-branching morphogenesis (Zhao et al., 1996). Expression of a dominant-negative mutant TGF-β IIR in transgenic mice blocks signaling by TGF-β isoforms in pancreatic acinar cells (Bottinger et al., 1997). TGF-β IIR null mutant mice show defects in the yolk sac hematopoiesis and vasculogenesis, resulting in an embryonic lethality at around 10.5 days of gestation (Oshima et al., 1996). The TGF-β IIR null mutant phenotype closely resembles that of TGF-β1 null mutant, thus suggesting that TGF-β IIR might likewise play an important role in regulating hematopoiesis and endothelial differentiation. During craniofacial development, TGF-β and its cognate receptor (IIR) may function in an autocrine/paracrine fashion to regulate the proliferation and apoptosis of enamel organ epithelial cells and CNC-derived dental mesenchyme, suggesting that TGF-β IIR is critical for controlling the size and stage of tooth development (Chai et al., 1999). In tandem, activin (a member of the TGF-β superfamily) and its receptor IIA play a critical role in regulating CNC-derived dental mesenchyme during incisor and mandibular molar tooth morphogenesis (Ferguson et al., 2001). Collectively, these studies demonstrate the crucial regulation and distinct activities of TGF-β receptors in the TGF-β signal transduction pathway during embryogenesis.
Smads are cytoplasmic mediators of TGF-β signaling
Until recently, little was known about the signaling molecules and intracellular substrates that mediate the TGF-β IR/IIR receptor hetero-complex responses to extracellular TGF-β ligand binding. Genetic studies in Drosophila uncovered the mothers against dpp (mad) gene as a maternal effector that constitutes a downstream component of signaling by decapentaplegic (Dpp), the Drosophila homologue of mammalian bone morphogenetic proteins (BMPs) (Raftery et al., 1995; Sekelsky et al., 1995). Overexpression of Mad protein complements Dpp deficiency, while Dpp fails to induce biological responses in Mad mutant flies (Newfeld et al., 1996; Wiersdorff et al., 1996). Sma2, Sma3, and Sma4 proteins, structurally related to Drosophila Mad, were also found in parallel studies of C. elegans development (Savage et al., 1996). These intracellular proteins are required for the function of Daf-4, a serine/threonine kinase receptor which responds to the C. elegans homologues of BMP-2 and BMP-4 (Estevez et al., 1993).
At least nine genes, homologous to mad and sma, have been identified in Xenopus, mouse, and human and shown to be components in signal transduction pathways downstream of serine/threonine kinase receptors (reviewed in Massagué, 1996, 1998; Heldin et al., 1997). The vertebrate homologues of Mad and Sma have been designated as Smad. Smads are molecules of relative mass 42-60 kDa, with two regions of homology at the amino and carboxy terminals, terminal Mad-homology domains MH1 and MH2, respectively, connected by more diverse, proline-rich linker sequences.
(1) Pathway-restricted Smads
Different members of the Smad family have distinct signaling functions. Smad1, 2, 3, and possibly 5 interact with and are phosphorylated by specific type I serine/threonine kinase receptors, and thereby act in a pathway-restricted manner. Smad2 and Smad3 are phosphorylated and translocated to the nucleus after stimulation by TGF-β (Fig. 1) (Eppert et al., 1996; Zhang et al., 1996; Nakao et al., 1997a,b) or activin (Y Chen et al., 1996). Smad2 and Smad3 are very similar in their structures. It is possible that there may be some redundancy in the functional activity between these two family members. Smad1 is phosphorylated and translocated into the nucleus after stimulation by BMP-2 or BMP-4 (Hoodless et al., 1996; Liu et al., 1996; Kretzschmar et al., 1997a). Like Smad1, Smad5 induces ventral mesoderm in Xenopus (Suzuki et al., 1997). The recently identified Smad9 (Watanabe et al., 1997) is structurally similar to Smad1 and Smad5, indicating that these molecules may also be involved in BMP signaling. The pathway-restricted Smads are direct substrates of type I receptor kinases at their serine residues in the consensus C-terminal SSXS motif (Macías-Silva et al., 1996; Abdollah et al., 1997; Kretzschmar et al., 1997a,b; Souchelnytskyi et al., 1997). The interaction between type I receptors and pathway-restricted Smads is transient: Smads are released from receptors after phosphorylation. Smad2, when mutated at the three serine residues in the SSXS motif, binds stably to the receptor and has a dominant-negative effect (Macías-Silva et al., 1996; Abdollah et al., 1997; Souchelnytskyi et al., 1997). The activities of Smad3 and Smad4 are regulated by TGF-β receptors. Smad3 but not Smad4 is phosphorylated and associated with TGF-β ligand-bound receptor complex (Zhang et al., 1996). The efficient phosphorylation of Smad3 by the type I receptor kinase in vitro suggests that Smad3 may be a direct kinase target of the TGF-β receptor complex.
(2) Common-mediator Smad
The action of Smad4 differs from those of other members of the Smad family. After ligand stimulation and phosphorylation of pathway-restricted Smads, Smad4 forms hetero-oligomers with pathway-restricted Smads (Lagna et al., 1996; Zhang et al., 1996; Kretzschmar et al., 1997a,b; Wu et al., 1997), which in turn translocate into the nucleus and selectively effect transcriptional responses (Fig. 1). In mammalian cells, Smad4 forms complexes with Smad2 and Smad3 after activation of TGF-β (X Chen et al., 1996; Y Chen et al., 1996; Lagna et al., 1996; Wu et al., 1997), whereas after activation of BMP type I receptors, it forms complexes with Smad1 (Lagna et al., 1996; Kretzschmar et al., 1997a,b), and possibly with Smad5 and Smad9. Smad4, which lacks the C-terminal SSXS motif, does not bind to, nor is it phosphorylated by, TGF-β or BMP receptors.
(3) Inhibitory Smads
Smad6 and Smad7 diverge structurally from other members of the Smad family (Hayashi et al., 1997; Imamura et al., 1997; Topper et al., 1997; Massagué, 1998), they function as inhibitors of TGF-β, activin, and BMP signaling. In particular, Smad6 inhibits BMP/Smad1 signaling by specifically competing with Smad4 for binding to receptor-activated Smad1, without interfering with the receptor-mediated phosphorylation of Smad1 (Hata et al., 1998). Smad7 associates stably with the TGF-β receptor complex, but is not phosphorylated upon TGF-β stimulation. Smad7 inhibits TGF-β-mediated phosphorylation of Smad2 and Smad3 (Fig. 1). Because transcription of the inhibitory Smad gene is induced by stimulation of TGF-β (Nakao et al., 1997b), inhibitory Smads may produce autoregulatory negative feedback in the signal transduction of the TGF-β superfamily.
Collectively, these studies show that Smads are intracellular mediators for the TGF-β signaling pathway. Furthermore, Smad2, Smad3, and Smad4 are not only present in the cytoplasm but also concentrated in the nucleus of transfected cells, where they are thought to play a role in transcriptional regulation of TGF-β responsive genes (Derynck, 1994; Wu et al., 1997; Massagué, 1998, 2000). All Smads have transcriptional activity, as both activator and repressor (Massagué and Wotton, 2000; Moustakas et al., 2001). Various mechanisms have been proposed to control the trafficking of Smads between cytoplasm and nucleus (Massagué, 2000). Once inside the nucleus, receptor-regulated Smad binds with Smad co-factors and transcriptional co-activators/co-repressors to regulate the target gene expression specifically. It is important to point out that a specific cellular response to TGF-β signaling is largely dependent on the cellular context, but not on TGF-β itself (Massagué, 2000).
Targeted mutagenesis in mice has yielded valuable insights into the biological functions of Smads gene family. Both Smad2 and Smad4 are critical for gastrulation process. Smad2 null mutant embryos are smaller than their littermates, lack the extra-embryonic portion of the egg cylinder, fail to establish the anterior-posterior polarity of body plan, and die before E8.5 (Nomura and Li, 1998; Waldrip et al., 1998). Smad4 null mutant embryos fail to form mesoderm and die before E7.5 (Sirard et al., 1998). Smad3 null mutant mice develop normally. Adult Smad3 null mutant mice have impaired mucosal immunity, diminished T-cell responsiveness to TGF-β, and form metastatic colorectal cancer (Zhu et al., 1998; Yang et al., 1999). Because of the high homology between Smad3 and Smad2 (> 90%), it has been speculated that the shorter form of Smad2 (alternatively spliced Smad2 without exon 3) may function as Smad3 in transducing TGF-β signaling and, thus, can compensate for the Smad3 null mutation (Yagi et al., 1999). Recently, Smad3 has been shown to play a more important role in TGF-β-mediated pathogenetic events and to be less involved during embryonic development (Ashcroft et al., 1999).
Significantly, targeted Smad mutation experiments have demonstrated the importance of proper Smad gene dosage in carrying out TGF-β signaling, since several Smad heterozygous mutants yield functional haploinsufficiency phenotypes (for review, see Weinstein et al., 2000). Specifically, Smad2 heterozygous mutation results in craniofacial defects, such as mandibular hypoplasia and cyclopia, indicating a possible cranial neural crest defect (Nomura and Li, 1998). Functional haploinsufficiency of Smad2 affects TGF-β/activin-mediated tooth development (Ferguson et al., 2001; Ito et al., 2001). Since TGF-β signal transduction is not subject to a great deal of amplification between the cell membrane where the signal is received and the nucleus where the target gene is regulated, it is of pivotal importance that we appreciate the biological significance of the concentration of TGF-β signaling Smad, because its expression level can clearly affect the effectiveness of TGF-β signaling. Furthermore, from a functional point of view, recent studies have revealed that receptor-regulated and inhibitory Smads may have differential activities in regulating TGF-β-mediated cell proliferation and death, thus providing further supportive evidence that Smads are critical factors in orchestrating TGF-β signaling during embryogenesis (Ito et al., 2001, 2002).
(II) The Cranial Neural Crest
The vertebrate neural crest is a pluripotent cell population derived from the lateral ridges of the neural plate during early stages of embryogenesis. Neural crest cells disperse from the dorsal surface of the neural tube and migrate extensively through the embryo, giving rise to a wide variety of differentiated cell types. The differentiation of neural crest cells along multiple distinctive pathways and their contribution to the formation of a particular tissue or organ have generated considerable interest in developmental biology (Romer, 1972; Noden, 1983; Tan and Morriss-Kay, 1986; Bronner-Fraser, 1993; LaBonne and Bronner-Fraser, 1999). Progress has been made in recent years toward our understanding of how this important population of pluripotent cells is initially established in the early embryo, and how genetic and epigenetic mechanisms mediate their subsequent lineage segregation, differentiation, and final contribution to a particular phenotype (see review by Le Douarin et al., 1994; Shah et al., 1996; Shah and Anderson, 1997; LaBonne and Bronner-Fraser, 1999).
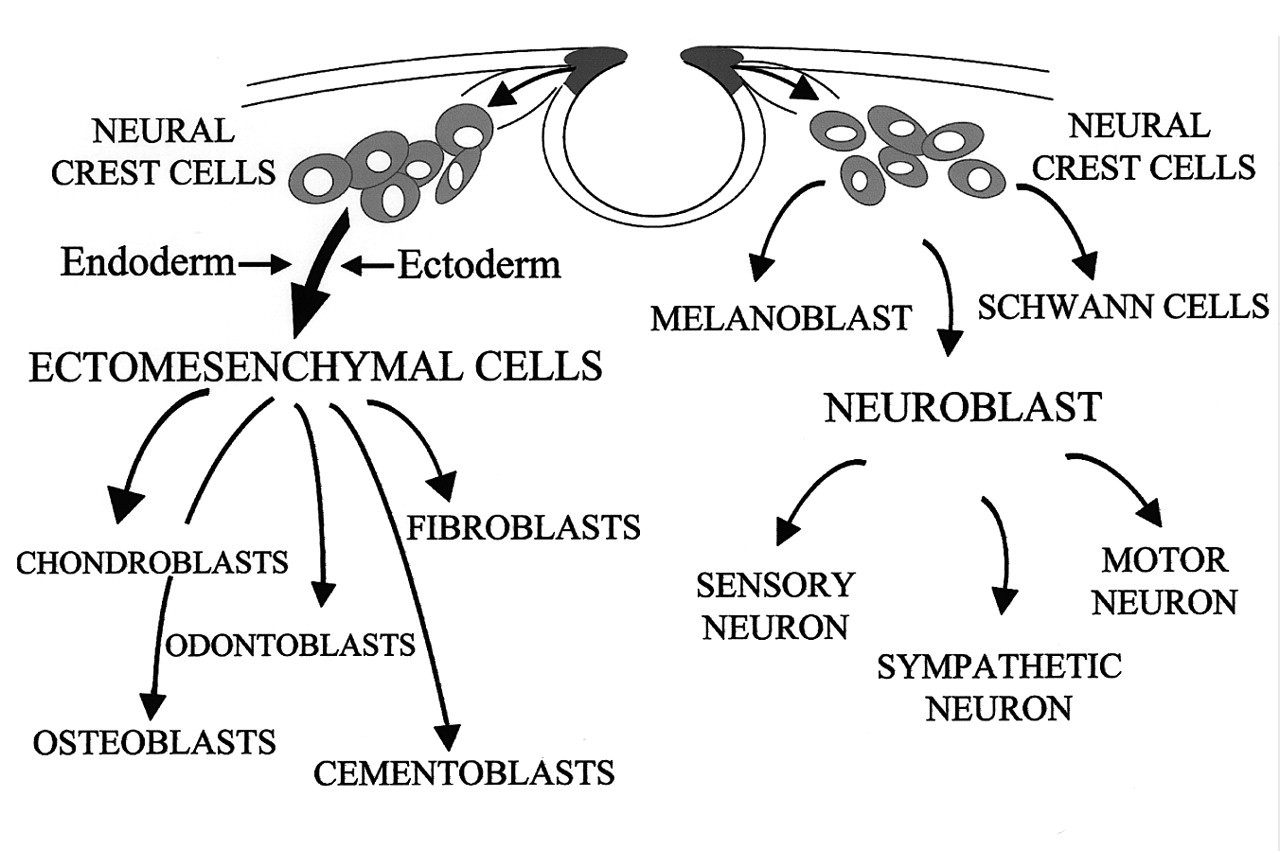
During craniofacial development, neural crest cells migrate ventro-laterally as they populate the branchial arches. The proliferative activity of these crest cells produces the discrete swellings that demarcate each branchial arch. As these ectodermally derived cells migrate, they contribute extensively to the formation of mesenchymal structures in the head and neck. Cell-labeling studies have demonstrated that neuroectoderm cells of rhombomeres 1-3 (r1-3) in the forming posterior midbrain and anterior hindbrain transform into cranial neural crest cells which migrate into the first branchial arch and thereafter reside within the maxillary and mandibular prominences (Osumi-Yamashita et al., 1990; Serbedzija et al., 1992; Bronner-Fraser, 1993; Selleck et al., 1993; Lumsden and Krumlauf, 1996). The migration of these rhombencephalic crest cells may be regulated by growth factor signaling pathways and their downstream transcription factors before they become committed to several different phenotypes, including progenitor tooth mesenchymal cells, osteoblasts, chondroblasts, and cranial nerve ganglia of the branchial arch (Fig. 2) (Noden, 1983, 1991; Lumsden, 1988; Graham and Lumsden, 1993; Le Douarin et al., 1993; Echelard et al., 1994; Imai et al., 1996; Trainor and Krumlauf, 2000). Recent studies provide strong supportive evidence that the CNC cells are developmentally ’plastic’-their fate is not pre-determined before they reach their final destination; rather, these progenitor cells must be instructed by signals from other tissues to generate skeletal elements of appropriate shape and size in the craniofacial region. Tissues which provide the instructive signaling for CNC fate specification include, but are not limited to, the isthmic organizer behind the midbrain region, cranial placode, and the branchial arch ectoderm and endoderm (Fig. 2) (Baker and Bronner-Fraser, 2001; Couly et al., 2002; Trainor et al., 2002).
CNC cell lineage studies using chick-quail chimeras (Noden, 1983), cell labeling with a vital dye such as DiI (Serbedzija et al., 1989, 1992), neural-crest-cell-specific antibodies (Tucker et al., 1984), and retroviral-mediated gene transfer (Poelmann and Gittenberger-de Groot, 1999) have significantly advanced our understanding of the migration and differentiation pathways of these multipotent stem cells during embryogenesis. However, a comprehensive cell lineage analysis of the mammalian neural crest cells, as they become terminally differentiated to become a particular phenotype, has been limited by the lack of a genetic marker permitting these cells to be followed indefinitely. A recently developed two-component genetic system for indelibly marking the progeny of neural crest cells has significantly improved our ability to analyze the fate of CNC during normal as well as abnormal craniofacial development (Chai et al., 2000).
(III) Fate Analysis of the Cranial Neural Crest
The proto-oncogene Wnt1 encodes a short-range signal and is expressed only during development of the central nervous system (Wilkinson et al., 1987; McMahon et al., 1992; Echelard et al., 1994). Wnt1 expression is initiated at neural plate stages throughout the presumptive midbrain, and then becomes rapidly restricted to a tight circle lying just anterior of the mid/hindbrain isthmus at the time of neural tube closure. Spontaneous and targeted mutations of Wnt1 result in the loss of the midbrain and lead to a secondary loss of anterior hindbrain (McMahon and Bradley, 1990; Thomas and Capecchi, 1990; Thomas et al., 1991; McMahon et al., 1992; Augustine et al., 1993). Significantly, cranial and spinal ganglia and skeletogenic neural crest cells in the branchial arches are all derived from Wnt1 expressing precursor cells in CNS. Transgenic lines expressing β-galactosidase under the control of Wnt1 promoter demonstrate staining in the CNS, identical to the expression pattern of the endogenous Wnt1 gene, and show staining in the population of neural crest initially emigrating away from the neural tube (Echelard et al., 1994). Thus, Wnt1 transgene expression provides a potential new tool for analysis of neural crest development.
To use Wnt1 transgene expression as a marker to follow the migration and differentiation of neural crest cells, the transgene must be active throughout embryogenesis and beyond. With this Wnt1-lacZ transgene, however, β-galactosidase-positive cells are not seen in later embryos, thereby accurately reflecting the cessation of Wnt1 gene expression in the neural crest progeny. Without lacZ expression in later embryos, this Wnt1-lacZ transgenic line cannot be used to follow the differentiation of neural crest cells. To solve this problem and to be able to follow the migration and differentiation of CNC cells indefinitely, researchers have taken advantage of the cre/lox system. The Wnt1-cre transgene mediates DNA recombination after being crossed with the ROSA26 conditional reporter (R26R) transgene. R26R exhibits constitutive β-gal expression in all cells when activated by ubiquitously expressed cre, and is thus ideal for monitoring cre-mediated expression and cell lineage analysis in both developmental and post-natal times (Soriano, 1999). By utilizing the Wnt1 promoter, cre expression is restricted to the precursors of the neural crest. Consequently, the progeny of the neural crest is marked indelibly with β-gal during embryogenesis. Recent studies using this two-component genetic system have systematically followed the dynamic contribution of neural crest cells during embryogenesis (Chai et al., 2000; Jiang et al., 2000, 2002; Ito et al., 2002; Chai and Slavkin, 2002). More importantly, this genetic approach provides the opportunity to integrate the analysis of the fate and function of mammalian neural crest with mouse molecular genetics, thus elucidating the molecular mechanism of neural-crest-related congenital malformations (Brault et al., 2001).
(IV) TGF-β Regulates the Expression of Transcription Factors to Determine the Fate of Cranial Neural Crest Cells
Recent studies have focused on how growth factors influence the fate of multipotent progenitor cells, such as the neural crest, during embryogenesis. Members of the TGF-β superfamily of growth factors are expressed at sites where neural crest cells are committed to form a particular cell type. Alternative neural crest cell fates are instructively promoted by TGF-β superfamily members, of which BMP signaling promotes neurogenesis by inducing MASH1 expression, while TGF-β signaling favors smooth-muscle differentiation (Shah et al., 1996). Furthermore, the function of TGF-β in regulating neural crest cell differentiation is sensitive to the TGF-β expression level, at which TGF-β may promote alternative cell fates or induce apoptosis (Hagedorn et al., 2000). During early mouse craniofacial development, the presence of TGF-β subtypes is obvious in CNC-derived mesenchyme during critical epithelial-mesenchymal interactions related to the formation of various organs (Lumsden, 1984; Hall, 1992; Chai et al., 1994; Lumsden and Krumlauf, 1996). Targeted null mutation of TGF-β2 or haploinsufficiency of Smad2 results in a wide range of developmental defects, including craniofacial malformations such as small mandible, dysmorphic calvaria, and cleft palate (Sanford et al., 1997; Nomura and Li, 1998). Significantly, many affected tissues have neural-crest-derived components and simulate neural crest deficiencies; thus, TGF-β signaling may provide significant instructive information to specify the fate of CNC during early craniofacial development.
Members of the TGF-β superfamily regulate the expression of transcription factors to influence cell fate decisions instructively during embryogenesis (Shah et al., 1996; Dorsky et al., 2000). For example, the expression patterns of TGF- β 2 and transcription factor Msx1 have significant overlaps during early tooth development, when CNC-derived cells become specified to form dental mesenchyme (http://bite-it.helsinki.fi/ and references therein) and suggest an epistatic relationship between these two genes.
Msx1 is strongly expressed in the CNC cells at the onset of migration and plays a critical role in regulating epithelial-mesenchymal interactions during embryogenesis (Hill et al., 1989; Robert et al., 1989; Lyons et al., 1992). Recent studies have shown that Msx1 is critical in the maintenance of progenitor cells in the undifferentiated state and may induce cellular pluripotency (Song et al., 1992; Akimenko et al., 1995; Simon et al., 1995; Woloshin et al., 1995; Odelberg et al., 2000). The Msx gene may regulate the expression of cell cycle regulatory molecules, thus controlling the progression of the cell cycle by regulating progenitor cell proliferation, differentiation, and apoptosis during embryogenesis (Gomes and Kessler, 2001; Hu et al., 2001). During craniofacial development, Msx1 is expressed continuously as CNC-derived ectomesenchyme differentiates into bone and teeth (MacKenzie et al., 1991a,b, 1992). Targeted null mutation of Msx1 resulted in craniofacial abnormalities that include small mandibles and maxilla, arrested tooth development at the bud stage, and cleft palate, indicating a possible deficiency of CNC cells (Satokata and Maas, 1994; Houzelstein et al., 1997). Although there is no indication that heterozygous mutation of Msx1 is associated with developmental defects in mice, it is well-documented that haploinsufficiency of MSX1 in humans results in congenital malformations, such as selective tooth agenesis and facial clefting, and suggests a possible CNC defect (Vastardis et al., 1996; Hu et al., 1998; van den Boogaard et al., 2000; Jumlongras et al., 2001). Thus, the level of Msx1 expression is critical for the proper regulation of the fate of CNC cells during craniofacial development.
By using the two-component genetic system to mark the progenies of CNC cells, a recent study provides the first direct evidence of CNC cell deficiency within the affected craniofacial structures of Msx1 null mutant mouse embryos. The deficiency of CNC results from reduced mitotic activity which is inhibited by an elevated CDK inhibitor expression within the odontogenic mesenchyme. Elevated CDK inhibitor activity interferes with proper phosphorylation of the Rb proteins and thus disrupts the progression of the cell cycle, interferes with proper CNC differentiation, and ultimately results in apoptosis of these progenitor cells and failure of tooth morphogenesis. Attenuation of the CDK inhibitor in Msx1 null mutant mandibular explants rescues tooth development at the cap stage, demonstrating the critical function of Msx1 in regulating CNC cell-cycle progression during tooth formation (Han et al., manuscript under review). Collectively, these studies demonstrate the critical function of Msx1 in regulating cranial neural crest cell fate determination during craniofacial development.
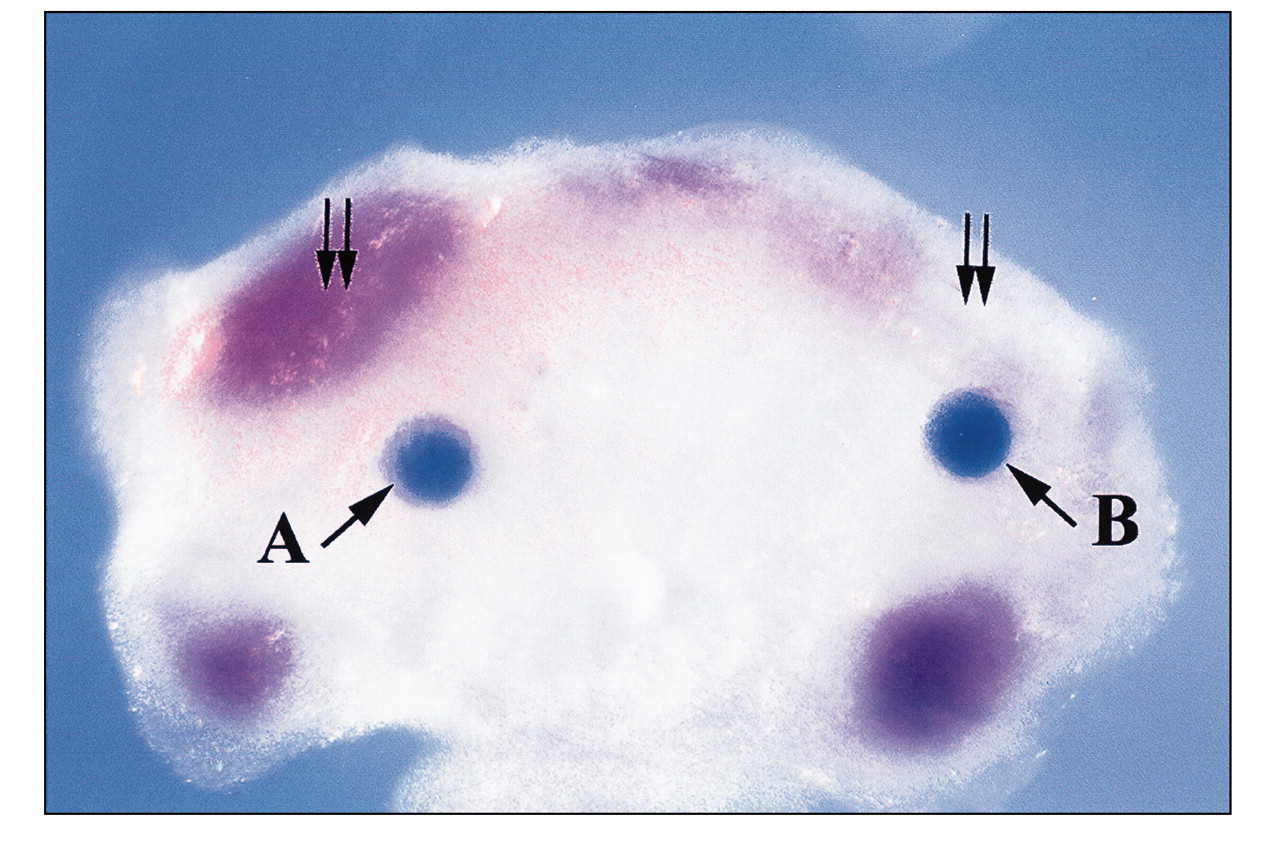
The genetic interaction between TGF-β signaling and the Msx gene plays an important role in the regulation of embryogenesis. Specifically, TGF-β down-regulates the expression of Msx1 and affects cell fate determination in limb development (Ganan et al., 1996). Altered Msx gene expression results in premature differentiation of the interdigital mesoderm into connective tissue in the developing autopod, thus preventing digit separation (Ganan et al., 1998). Overexpression of TGF- β suppresses the transcriptional activity of the Msx1 gene during palatal development (Nugent and Greene, 1998), while compound heterozygous mutation of Smad2 and Smad3 results in elevated Msx1 expression (Ito et al., unpublished data). Mutations of Msx1 or TGF- β 3 have been associated with cleft palate in humans (Hu et al., 1998; Lidral et al., 1998). During the first branchial arch morphogenesis, TGF-β is mainly expressed in the ectoderm, while Msx1 expression is exclusively in the CNC-derived ectomesenchyme (MacKenzie et al., 1991a,b, 1992; Chai et al., 1994). Significantly, the expression of Msx1 is down-regulated by overexpression of TGF-β2 in the CNC-derived ectomesenchyme of the molar tooth region, suggesting an important biological function of TGF-β-mediated Msx1 expression during the CNC fate determination (Fig. 3). Taken together, these studies offer supportive evidence of an epistatic relationship between TGF-β signaling and the expression of Msx1 in regulating epithelial-mesenchymal interaction during organogenesis. Given the fact that the fate of the CNC is sensitive to the Msx1 expression level, future studies exploring the hierarchy of TGF-β signaling and its regulatory function on the expression of transcription factors, such as the Msx gene, will provide us with a better understanding of the molecular regulation of early embryogenesis.
There are other examples of neural crest cell fate specification induced by specific transcription factors which are regulated by members of the TGF-β superfamily. The transcription factor dHAND is an important member of the network of transcriptional regulators involved in neural-crest-derived sympathetic neuron development and is a downstream effector of BMP signaling (Howard et al., 2000). Both dHAND and eHAND are members of the basic helix-loop-helix (bHLH) family of transcription factors and regulate determination and differentiation of progenitor cells as they give rise to skeletal myocytes, neurons, and hematopoietic cells (Jan and Jan, 1993; Olson and Klein, 1994; Lee et al., 1995; Shivdasani et al., 1995; Thomas et al., 1998; te Welscher et al., 2002). Significantly, the first branchial arch ectoderm secretes a signal to stimulate the mesenchymal expression of dHAND which can regulate Msx1 expression, thus suggesting that this signaling pathway plays an important regulatory role during craniofacial morphogenesis. Complete disruption of this molecular signaling pathway leads to growth failure of the branchial arches due to cell death, while partial disruption results in defects of branchial arch derivatives, similar to those seen in CATCH-22 syndrome, including cardiac and facial defects (Wilson et al., 1993; Thomas et al., 1998).
Using tooth morphogenesis as an example, recent studies have significantly advanced our understanding of the molecular ’rules’ for cell determination, cell-cell recognition, and heterologous tissue-tissue interactions during embryogenesis. For example, during initial first branchial arch development, FGF8 growth factor is critical for the specification of oral epithelium. The function of FGF8 appears to regulate the expression of two specific Lim-homeobox domain genes, Lhx6 and Lhx7, within CNC-derived dental mesenchyme (Grigoriou et al., 1998). Interestingly, CNC-derived ectomesenchyme is mainly localized or restricted to the position immediately adjacent to the lining oral epithelium of the first branchial arch, as compared with the suboral region (Chai et al., 2000). This CNC distribution pattern significantly overlaps with the expression of Lhx6 and Lhx7, suggesting that these two homeobox genes may have critical functions in directing CNC cells to reach their destination. During the specification of the initiation of tooth development, antagonistic signaling between the gradients of FGF and BMP growth factors may selectively regulate the expression of certain transcription factors within CNC-derived dental mesenchyme and, consequently, determine the sites of tooth bud formation (Neübuser et al., 1997). The significance of the antagonistic signaling between FGF and BMP ligands is further demonstrated by the inhibition of BMP functions. For example, modifications in these growth factor gradients and their consequence can change tooth form from incisor to molar. This is demonstrated, for example, when such gradient changes result in ectopic Barx-1 expression in the distal, presumptive incisor mesenchyme, and produce transformation of tooth form from incisor to molar (Tucker et al., 1998). Both FGF and BMP growth factor gradients are critical molecules for specifying the initiation sites for tooth formation as well as the CNC-derived dental mesenchyme specificity to become incisor-form vs. molar-form tooth germs.
Classic tissue recombination experiments have also indicated that early dental ectoderm may determine tooth formation (Mina and Kollar, 1987; Lumsden, 1988). This is not to say, however, that the CNC-derived ectomesenchyme will just be passively instructed by oral ectoderm during the specification of tooth development, since it has been shown that the CNC-derived cells are also instructive for the initiation and patterning of tooth development (Thomas et al., 1997; Chai et al., 1998). To understand this complex process during the determination of tooth development, it is critical that we appreciate the evidence within a chronological perspective, since the sequence in time and position of these informative epithelial-mesenchymal interactions, which determine the initiation and specification of tooth formation, continues throughout all stages of tooth development (the dental lamina, the bud, the cap, the bell, and tooth crown stages). Furthermore, all of the oral ectodermal signaling molecules (such as BMPs, FGFs, and Shh), which have been shown to be critical regulatory molecules for the initiation of tooth formation, are also critical for the initiation of other forming organs, such as limb, hair, and mammary gland development. A remaining critical question is: How do comparable signaling molecules provide the unique specificity required for the initiation of epidermal organs such as tooth morphogenesis? So far, all evidence supports the hypothesis that a ’combinatorial process’ provides the specificity for these unique interactions between the oral ectoderm-derived signaling molecules and the specific transcriptional regulators in the CNC-derived dental mesenchyme (such as Msx, Pax, Barx, and Dlx transcription factor genes) that result in tooth morphogenesis.
Members of the transforming growth factor-β (TGF-β) family have been implicated as the key regulators during the transition from dental lamina to bud stage development. Their function may involve both the induction of a signaling center in the forming bud stage dental epithelium and the control of cell proliferation (Vainio et al., 1993; Chai et al., 1994, 1999; Bei and Maas, 1998; Ferguson et al., 1998). In particular, because both BMP4 and activin βA are present in the CNC-derived dental mesenchyme, their ability to induce the tooth development to advance into the bud stage may help to explain the shift in tooth-inducing potential from oral ectoderm to ectomesenchyme, as was demonstrated in an early tissue recombination experiment (Mina and Kollar, 1987). TGF-β in the early dental lamina functions mainly as an anti-mitotic factor and may provide an intrinsic control to the proliferation stimulators such as Shh and PDGF-A (Chai et al., 1994, 1998, 1999; Cobourne et al., 2001).
Conclusion
Elucidating the molecular pathways and mechanisms regulating the fate of the cranial neural crest is fundamental to our understanding of the pathogenesis of numerous congenital syndromes involving the development of craniofacial structures. TGF-β signaling plays a critical role during craniofacial development by regulating the specification of CNC cells. The transduction of TGF-β signal from the cell surface to the nucleus is subject to multiple steps of regulation, ranging from a TGF-β antagonist outside the cell to co-factors in the nucleus. These regulatory molecules and TGF-β signaling intermediates (e.g., TGF-β receptors and Smads) form a signaling regulation network through which the temporal and spatial specification of TGF-β signaling is tightly controlled. Consequently, the multipotent CNC cells are instructed to become specific cell types during embryogenesis. Needless to say, TGF-β signaling is only one of the many regulatory molecules involved in the specification of CNC cells during craniofacial development. A clear understanding of this signaling network and the possible interactions with other signaling networks will begin to address the biological significance of the concerted action between growth and transcription factors in regulating normal development and disease. Ultimately, application of this knowledge will help us to reduce the pain and suffering associated with human birth defects.
TGF-β ligand binding to the heteromeric receptor complex induces phosphorylation of the type I receptors by the constitutively active type II receptors. Receptor activation then results in phosphorylation of receptor-associated Smads (Smad2 and Smad3). The phosphorylated Smads may then cooperate with Smad4, which is not associated with the receptor or phosphorylated by it. The cooperativity of receptor-associated Smads with Smad4 may be a general prerequisite for receptor signaling. The activated, heteromeric Smads complex will translocate into the nucleus to function as a transcriptional regulator, which can regulate the expression of transcription factors. Smad7 associates stably with the TGF-β receptor complex, but is not phosphorylated upon TGF-β stimulation. TGF-β-mediated phosphorylation of Smad2 and Smad3 is inhibited by Smad7, indicating the antagonistic effect of Smad7 in regulating the TGF-β signaling pathway. Differentiation of cranial neural crest cells. As CNC cells migrate into the craniofacial region, these ectomesenchymal progenitors may give rise to an array of tissue types, such as odontoblasts, chondroblasts, osteoblasts, etc. Both ectoderm and endoderm of the branchial arch provide signaling instructions for the fate specification of these progenitor cells. CNC cells also contribute to the formation of neural tissues, such as sensory neurons and cranial nerve ganglia. The expression of Msx1 is inhibited by TGF-β signaling. E11 mandibular explant (cultured for 2 days in serumless, chemically defined medium) shows proper Msx1 expression (double arrow) in the future molar tooth region adjacent to the BSA-bearing (0.1% bovine serum albumin) bead 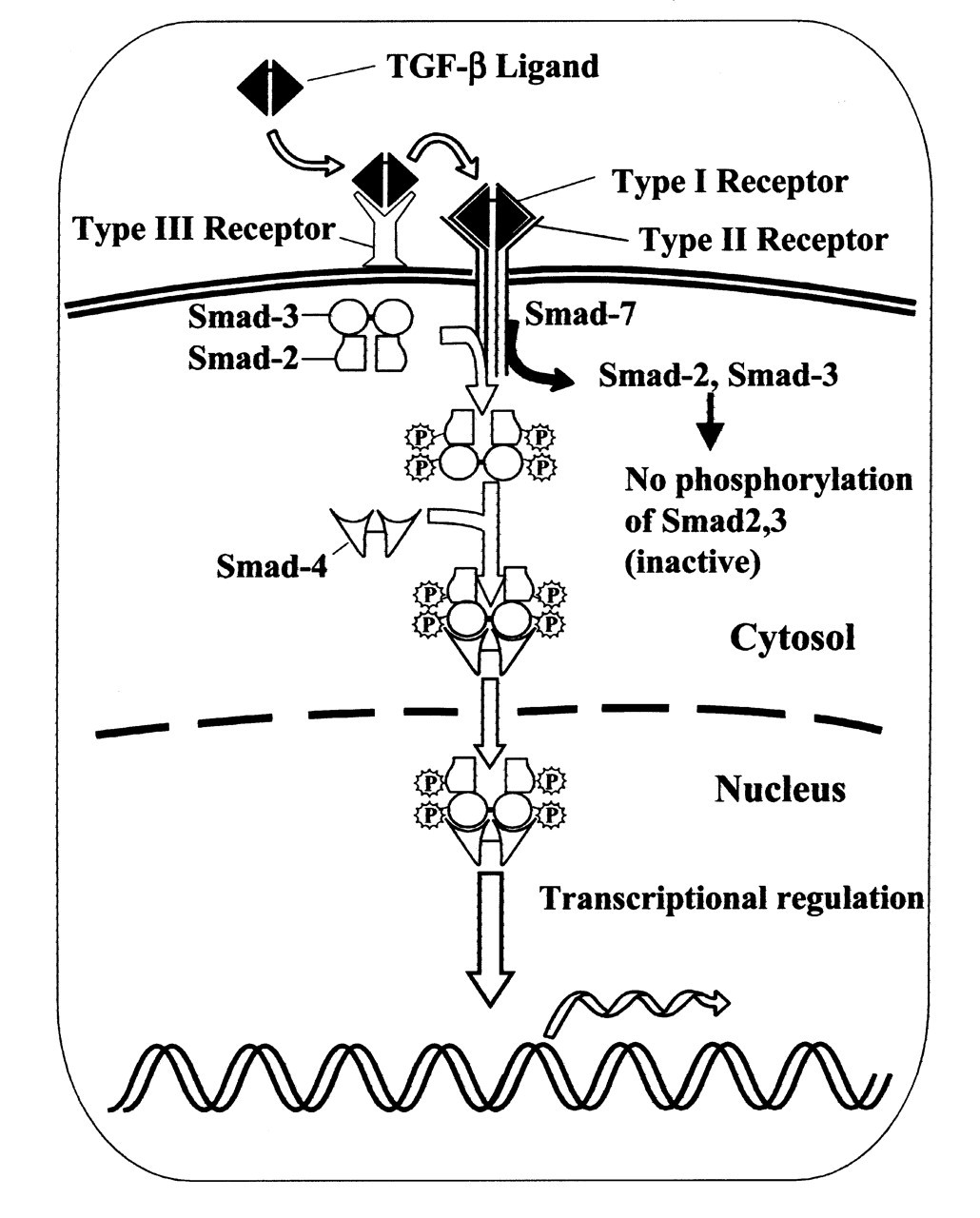
Footnotes
Acknowledgements
The author thanks Harold Slavkin and Malcolm Snead for their encouragement and Pablo Bringas, Jr. for his untiring effort and continued support over the years. Dr. Xu’s assistance is greatly appreciated. Studies in Yang Chai’s lab are supported by grants from the National Institute of Dental and Craniofacial Research, NIH (DE12711 and DE12941).