
Abstract
Pemphigus is a group of potentially life-threatening diseases characterized by cutaneous and mucosal blistering. There is a fairly strong genetic background to pemphigus with linkage to HLA class II alleles. Certain ethnic groups, such as Ashkenazi Jews and those of Mediterranean origin, are especially liable to pemphigus. Pemphigus vulgaris (PV), the most common and important variant, is an autoimmune blistering disease characterized by circulating pathogenic IgG antibodies against desmoglein 3 (Dsg3), about half the patients also having Dsg1 autoantibodies. Oral lesions are initially vesiculobullous but readily rupture, new bullae developing as the older ones rupture and ulcerate. Biopsy of perilesional tissue, with histological and immunostaining examinations, is essential to the diagnosis. Serum autoantibodies to either Dsg1 or Dsg3 are best detected by both normal human skin and monkey esophagus or by enzyme-linked immunosorbent assay (ELISA). Before the introduction of corticosteroids, pemphigus vulgaris was typically fatal mainly from dehydration or secondary systemic infections. Current treatment is largely based on systemic immunosuppression using systemic corticosteroids, with azathioprine, dapsone, methotrexate, cyclophosphamide, and gold as adjuvants or alternatives, but mycophenolate mofetil and intravenous immunoglobulins also appear promising.
Introduction
Pemphigus is a group of potentially life-threatening autoimmune mucocutaneous diseases characterized by epithelial blistering affecting cutaneous and/or mucosal surfaces, the term being derived from the Greek Pemphix (bubble or blister). Pemphigus affects 0.1-0.5 patients per 100,000 population per year (Ahmed et al., 1980; Becker and Gaspari 1993).
Oral lesions of pemphigus are seen in up to 18% of patients at dermatology out-patient clinics (Ramirez-Amador et al., 2000), but despite the frequency of oral involvement, and novel therapeutic approaches (Stanley, 2000), there are surprisingly few recent studies of either the oral manifestations of pemphigus or their management (Mashkilleyson and Mashkilleyson, 1988; Lamey et al., 1992; Robinson et al., 1997; Scully et al., 1999; Sirois et al., 2000), and delays in diagnosis are still common (Sirois et al., 2000).
Pemphigus has been reviewed in the oral literature in the past decade (Eversole, 1994; Weinberg et al., 1997), but several advances in the understanding of the etiopathogenesis, pemphigus variants, and management warrant an update.
Epithelial Biology
An elementary knowledge of the biology of the oral epithelium is crucial to the understanding of pemphigus. The oral epithelium is a complex structure consisting of a range of cells, mainly keratinocytes, adherent to each other by desmosomes, and via hemidesmosomes, to an epithelial basement membrane and thereby to the underlying lamina propria. Each component is itself complex and consists of several proteins with important functions—not least the adherence of cells to adjacent structures, and cell-cell recognition and signaling.
Cell-basement membrane contact is largely via hemidesmosomes, which have a complex structure and come into contact with the superficial part of the basement membrane (the lamina lucida). Cell-cell contact is via occludens (tight junctions), adherens (desmosomes and adhesion plaques), and nexus junctions (gap junctions), each having a complex structure.
Desmosomes
Desmosomes guarantee the integrity of the epithelia, by functioning both as an adhesive complex and as a cell-surface attachment site for the keratin intermediate filaments of the cytoskeleton. Desmosomes are adhesion proteins that contain a series of proteins, particularly desmogleins and desmocollins—glycoproteins of the cadherin supergene family which link to cytokeratins via desmoplakins and plakoglobin (Buxton and Magee, 1992). Cadherins are composed of an extracellular domain involved in calcium-dependent binding to adjacent cells, a transmembrane domain, and an intracellular domain that binds to catenins and thence to actin (Gumbiner and McCrea, 1993).
Oral epithelium
Oral epithelium is similar to skin but differs in several essentials, not least in that desmosomal components differ somewhat; for example, the cadherin-type adhesion molecules desmoglein 1 (Dsg 1) and Dsg 3 both are expressed in skin but oral epithelium expresses predominantly only Dsg 3 (Shirakata et al., 1998), the 130-kd molecule. This has consequences in terms of disease manifestations, as discussed below, as well as in antibody detection.
The epithelium thus has a complex structure (Fig. 1), and an array of molecules is required for epithelial integrity and health (Uitto and Pulkkinen, 1996; Lin et al., 1997; Cozzani et al., 2000). Damage to the intercellular area leads to separation of the keratinocytes-acantholysis—which, though typical of pemphigus, may be seen in other conditions (Table 1).
Pemphigus and Variants
Pemphigus affects the skin and may also affect the mucosae of the mouth, nose, conjunctivae, genitals, esophagus, pharynx, and larynx; it is found mainly in middle-aged and elderly patients. Pemphigus is a group of autoimmune disorders in which there is damage to desmosomes by antibodies directed against the extracellular domains of the cadherin-type epithelial cell adhesion molecules—the desmogleins (Dsg) (Nishikawa et al., 1996)—with immune deposits intra-epithelially, and loss of cell-cell contact (acantholysis), leading to intra-epithelial vesiculation.
Variants
There are several variants of pemphigus described (Table 2), with different autoantibody profiles and clinical manifestations.
Pemphigus vulgaris
Pemphigus vulgaris (PV) is the most common form and frequently involves the mouth (Weinberg et al., 1997; Scully et al., 1999). The main importance of PV is that it typically runs a chronic course, almost invariably causing blisters, erosions, and ulcers on the oral mucosae and skin. Before the introduction of corticosteroids, it was often fatal, mainly from dehydration or secondary systemic infections (Ahmed and Moy 1982; Robinson et al., 1997; Scully et al., 1999). The main antigen in pemphigus vulgaris is Dsg 3 (Amagai et al., 1992), whereas that in pemphigus foliaceus is Dsg 1 (Amagai et al., 1995). However, 50% of PV patients also have autoantibodies to Dsg1, and the proportion of Dsg1 and Dsg3 antibodies appears to be related to clinical severity (Harman et al., 2000a). Those PV which are predominantly oral have only Dsg3 antibodies (Harman et al., 2001). Typically, an individual patient develops a single variant of pemphigus, though cases have been described of transition to another variant (Ishii et al., 2000), presumably through epitope spread, and the clinical manifestations of a single variant can change over time, as discussed below. This change may be related to changes in the proportions of Dsg1 and Dsg3 autoantibodies (Harman et al., 2001).
Paraneoplastic pemphigus
Apart from PV, the other important variant affecting the mouth is paraneoplastic pemphigus (PNP), usually associated with lymphoproliferative disease (Allen and Camisa, 2000), though one case with oral squamous carcinoma has been reported (Wong and Ho, 2000). Oral lesions may be the sole manifestation (Bialy-Golan et al., 1996) and have also been seen in all reported cases of paraneoplastic pemphigus (Laskaris et al., 1980; Anhalt et al., 1990; Camisa et al., 1992; Fullerton et al., 1992; Perniciaro et al., 1994).
Other variants
Oral lesions have been seen in less common pemphigus variants, especially in most cases with IgA pemphigus (intra-epithelial IgA pustulosis [IEAP]), and in some cases of pemphigus associated with inflammatory bowel disease (Stone, 1971; Lubach et al., 1984; Delfino et al., 1986; Fabbri et al., 1986; Schwermann et al., 1988; Prendiville et al., 1994). In contrast, other types of pemphigus—such as pemphigus foliaceus (PF) and erythematosus and pemphigus vegetans—only rarely affect the oral mucosae (Ahmed et al., 1980; Virgili et al., 1992; Mahé et al., 1996).
Since PNP has been recently reviewed (Allen and Camisa, 2000), the present review focuses on pemphigus vulgaris.
Etiopathogenesis
Pemphigus vulgaris is caused by autoantibodies against epithelial intercellular components, especially cadherins, and particularly desmogleins (Dsg 3 mainly but also Dsg1 in PV, Dsg 1 in PF), and, though the precise initiating environmental or lifestyle factor is usually unclear, there is a genetic basis to many cases.
Genetic background
Feel free to make corrections! There is a fairly strong genetic background to pemphigus vulgaris (PV), certain ethnic groups, such as Ashkenazi Jews and those of Mediterranean origin, being especially liable (Eller and Kest, 1941; Gellis and Glass, 1941; Pisanty et al., 1974). PV-IgG subclasses are detectable not only in patients but also in their first-degree relatives (Kricheli et al., 2000). Rare familial cases of PV have been reported (Starzycki et al., 1998).
HLA class II allele associations in PV are found with HLA-DR4 (DRB1*0402), DRw14 (DRB1*1041), and DQB1*0503 (Sinha et al., 1988; Ahmed et al., 1990, 1991, 1993; Matzner et al., 1995; Carcassi et al., 1996; Delgado et al., 1996, 1997; Lombardi et al., 1996; Nishikawa et al., 1996; Miyagawa et al., 1997; Mobini et al., 1997a; Loiseau et al., 2000). Asian alleles of the HLA-B15 family, including the allele B*1507, are significantly increased in comparison with normal controls, in Japanese patients with PV—but HLA class l alleles are not changed (Miyagawa et al., 2002). These HLA class II alleles appear critical to T-lymphocyte recognition of Dsg 3 peptides.
Two kinds of Dsg 3-derived peptides may be presented by HLA-DR according to the HLA polymorphism (DRB1*0402 or DRB1*14/0406). The DRB1*14/0406 PV-related molecules may be able to present Dsg 1 and Dsg 3 peptides, providing one explanation for cases of PV with combined responses to Dsg1 and to Dsg3 which are typified by a muco-cutaneous clinical phenotype (Loiseau et al., 2000).
Pathogenesis
Inevitably, any one or more of the desomosomal proteins can be defective or damaged, and this can result in loss of cell-cell adhesion leading to the clinical result of vesiculation, erosions, or ulcers which characterize pemphigus. Pemphigus vulgaris is an autoimmune disorder in which there is deposition of mainly IgG class antibodies intercellularly (Fig. 2) as well as damage to desmosomes by antibodies directed against the extracellular domains of cadherin-type epithelial cell adhesion molecules, particularly desmoglein 3 (Nishikawa et al., 1996). Since oral epithelium expresses largely Dsg 3 but skin expresses Dsg 1 as well as Dsg 3, damage by antibodies to Dsg 3, as in PV, results in oral lesions at an early stage, whereas skin integrity is maintained by Dsg 1; however, if Dsg 1 antibodies appear, cutaneous lesions appear to result and the disease tends to be more severe (Harman et al., 2000b) (Table 3).
The Dsg autoantibodies are of the IgG class and in active PV are predominantly IgG4 polyclonal antibodies but IgG1 while in remission (Bhol et al., 1995; Tremeau-Martinage et al., 1995). Dsg 1 autoantibodies are found in over 50% of cases of PV, and the frequency may differ with race, since they are found in a significantly greater proportion of patients of Indian origin than in white northern Europeans (Harman et al., 2000b).
There is direct evidence that autoantibodies against desmoglein 3 (Dsg3) are critical in the pathogenesis (reviewed by Kalish, 2000; Anhalt and Diaz, 2001; Kowalewski et al., 2001), since the transfer of PV serum IgG antibodies against Dsg3 into newborn mice induces a bullous skin disease resembling PV (Nishikawa et al., 1996; Ding et al., 1999; Hertl, 2000), and recombinant Dsg 1 and Dsg 3 absorb the antibodies that cause PV-like skin blisters in neonatal mice. Loss of tolerance against Dsg3 in both B- and T-cells appears important for the development of PV (Tsunoda et al., 2002). Dsg 3 forms from two types of small clusters on the nondesmosomal plasma membrane, i.e., either half-desmosome-like clusters with keratin intermediate filament (KIF) attachment or simple clusters without KIF attachment. PV-IgG-induced internalization of the nondesmosomal simple clusters of Dsg3 may represent the primary effects of PV-IgG on keratinocytes (Sato et al., 2000). Furthermore, there is evidence that the disease activity in general correlates with the level of serum autoantibodies, and in vivo injection produces the disease in monkeys and mice and human skin (Schiltz and Michel, 1976).
The precise mechanism of the acantholysis after pemphigus IgG binds to Dsg 3 on the cell surface is unknown but may involve proteinases (reviewed by Kalish, 2000; Anhalt and Diaz, 2001; Kowalewski et al., 2001). PV- IgG causes a transient increase in intracellular calcium and inositol 1,4,5-trisphosphate concentration, and subsequent activation of protein kinase C (PKC) in cell lines. The phosphatidylcholine (PC)-specific phospholipase C (PLC) pathway plays a major role in P-IgG-induced transmembrane signaling by causing long-term activation of PKC (Seishima et al., 1999). Plasminogen activation and apoptosis may also be involved (reviewed by Kalish, 2000; Anhalt and Diaz, 2001; Kowalewski et al., 2001). Late development of Dsg 1 antibodies in PV correlates with disease progression (Miyagawa et al., 1999); the appearance of antibodies against Dsg1 heralds involvement of skin and mucosae other than oral (Ding et al., 1997; Harman et al., 2000b).
Antigens other than desmoglein
Pemphigus autoimmunity may not be limited to antidesmoglein antibodies. Nondesmoglein antibodies induce pemphigus-like lesions in neonatal mice. Non-Dsg PV IgGs also cause gross skin blisters with PV-like suprabasal acantholysis and staining perilesional epithelium in a fishnet-like pattern, indicating that the PV phenotype can be induced without anti-Dsg 3 or anti-Dsg 1 antibody (Nguyen et al., 2000a,b).
Acantholytic autoantibodies target a novel human alpha 9 acetylcholine receptor regulating keratinocyte adhesion, a novel keratinocyte annexin-like molecule binding acetylcholine and termed pemphaxin (Nguyen et al., 2000b), and catenin (Mignogna et al., 2001). This is an area of controversy reviewed elsewhere (Kalish, 2000; Anhalt and Diaz, 2001; Kowalewski et al., 2001).
Cellular immunity in PV
Although the PV autoantibodies are pathogenic, the role of the cellular immune system in acantholysis is unclear. Although CD4 T-cells that recognize the extracellular domain of these desmosomal cadherins are present, any role for these is as yet undefined. There is only a sparse cellular infiltrate around the basement membrane zone, but autoreactive T-cell responses to Dsg 3 may be critical to the pathogenesis, since antibody production generally requires T-cell help, and the strong association with distinct HLA class II alleles (see above) suggests the involvement of CD4+ T-lymphocytes. These T-cells recognize epitopes of Dsg 3. Most of the T-cells are CD45RO (Hertl et al., 1998a,b), which help autoreactive B-lymphocytes to produce autoantibodies (Nishifuji et al., 2000). CD28-deficient mice (lacking a co-stimulatory signal for T-lymphocyte activation) are much more sensitive to the development of PV than are wild-type mice (Toto et al., 2000). T-cell recognition of epitopes of Dsg 3 may be crucial for the initiation and perpetuation of the production of Dsg 3-specific autoantibodies by B-lymphocytes (Hertl and Riechers, 1999). These autoreactive CD4+ T-cells preferentially produce TH2 cytokines such as interleukin 4 (IL-4), IL-6, and IL-10 (Wucherpfennig et al., 1995; Lin et al., 1997), but also TH1 cytokines such as gamma interferon (Hertl et al., 1998a,b; Hertl and Riechers, 1999). Autoantibodies of the TH2-dependent IgG4 subtype are preferentially seen in active PV, while autoantibodies of the TH1-dependent IgG1 subclass predominate upon remission. Healthy individuals who carry HLA class II alleles similar or identical to those highly prevalent in PV also develop autoreactive T-cell responses to Dsg 3. Autoreactive T-cells from PV patients produce both TH1 and TH2 cytokines, while autoreactive T-cells from healthy persons produce TH0 cytokines (Hertl and Riechers, 1999). Cytokines including interleukin-10 (Toto et al., 2000), interleukin-6, interleukin-15, and tumor necrosis factor-alpha (Ameglio et al., 1999) are probably involved in PV (Feliciani et al., 2000).
Possible Etiological Factors
Diet
The role of diet in the etiology of pemphigus is reviewed elsewhere (Brenner et al., 1998; Tur and Brenner, 1998), but garlic in particular may cause occasional cases of pemphigus (Ruocco et al., 1996a).
Drugs
Traditionally, drugs that are capable of inducing pemphigus are divided into two main groups according to their chemical structure: drugs containing a sulfhydryl radical (thiol drugs or SH drugs), such as penicillamine and captopril (Laskaris et al., 1980; Korman et al., 1991; Wolf et al., 1991; Laskaris and Satriano, 1993; Ruocco et al., 1996a; Shapiro et al., 2000); nonthiol or other drugs, the latter often sharing an active amide group in their molecule (Wolf and Brenner, 1994). Phenol drugs (Goldberg et al., 1999), rifampicin (Gange et al., 1976), diclofenac (Matz et al., 1997), captopril (Korman et al., 1991), and other ACE inhibitors (Kaplan et al., 1992; Ong et al., 2000) are occasionally implicated. Cosmetics have been implicated in the high prevalence of PV in Tunisia (Bastuji-Garin et al., 2002).
Viruses
The initiating factor in PV remains enigmatic but particularly in view of the apparently transmissible nature of some pemphigus variants (fogo selvagem), the role of viruses has been suggested (reviewed by Ruocco et al., 1996b). Most recently, attention has been directed toward the herpesviruses. Very occasionally, the onset of PV has been reported concurrently with (Takahashi et al., 1998), or following, herpesvirus infections, and the possibility of epitope spreading or molecular mimickry has been suggested as the pathogenesis (Goon et al., 2001). Herpesvirus DNA has been detected, by polymerase chain reaction, in peripheral blood mononuclear cells and skin lesions of patients with pemphigus (Tufano et al., 1999). Human herpesvirus 8 (HHV-8) DNA was detected in lesions of patients with PV, while all specimens of non-pemphigus blistering skin diseases were negative (Memar et al., 1997; Jang et al., 2000). When PCR products were sequenced, the sequences were almost identical to the prototypic sequence for HHV-8, and a few base-pair substitutions at 1086C-T and 1139A-C were detected, suggesting that HHV-8 might have trophism for pemphigus lesions (Jang et al., 2000). In contrast, others have failed to detect HHV-8 DNA in lesional skin of pemphigus vulgaris patients (Cohen et al., 1998; Bezold et al., 2000).
Other factors
A recent multicenter study at outpatient services of teaching hospitals in Bulgaria, Brazil, India, Israel, Italy, Spain, and the USA revealed lower numbers of smokers among patients with PV, higher exposure rates to pesticides, and a higher number of female patients who had been pregnant. These findings suggested that this may point to the contribution of estrogens in the disease process (Brenner et al., 2001).
Association with other disorders
Pemphigus vulgaris may occasionally be associated with other autoimmune disorders, such as rheumatoid arthritis, myasthenia gravis, lupus erythematosus, or pernicious anemia (Ahmed et al., 1980).
Oral Lesions
Oral lesions of PV are typically seen in adults, rarely in childhood (Laskaris et al., 1980). Oral lesions are common and early manifestations (Eversole et al., 1972) and typically run a chronic course, causing blisters, erosions, and ulcers (Fig. 3). However, the prevalence of oral involvement varies: One recent multicenter study in several countries showed that Bulgarian patients less frequently had oral mucous membrane lesions (66%) compared with Italian (83%) and Israeli (92%) patients (Brenner et al., 2001). Initially vesiculobullous, the oral lesions readily rupture, new bullae developing as the older ones rupture and ulcerate (Sciubba, 1996), and thus erosions and ulcers are the main features and are seen primarily in the buccal mucosa, palate, and lips (Pisanty et al., 1974; Meurer et al., 1977; Zegarelli and Zegarelli, 1977; Orlowski et al., 1983; Shah and Bilimoria, 1983; Sklavounou and Laskaris, 1983; Lamey et al., 1992; Kanwar and Dhar, 1995; Weinberg and Abitbol, 1995; Scully et al., 1999; Davenport et al., 2001). Ulcers heal slowly, but scarring is rare (Zegarelli and Zegarelli, 1977; Shklar et al., 1978). Gingival lesions are less common and usually comprise severe desquamative or erosive gingivitis, where bullae have ruptured to leave flaps of peeling tissue with red erosions or deep ulcerative craters mainly on the attached gingivae (Shklar et al., 1978; Markitziu and Pisanty, 1983; Orlowski et al., 1983; Barnett 1988). Desquamative gingivitis (DG) may be seen, but this is a term that denotes a particular clinical picture and is not a diagnosis in itself (Scully and Porter, 1997) (Fig. 4).
Diagnosis
Many disorders damaging epithelial adhesion molecules are of autoimmune etiology, may have systemic manifestations (Eversole, 1994; Weinberg et al., 1997), and can be difficult to differentiate clinically. Therefore, without further investigation, it can be difficult if not impossible to determine the molecular basis of vesiculobullous, erosive, or ulcerative disorders affecting the oral mucosa or gingivae. Something as apparently homogeneous as desquamative gingivitis is thus a catch-all term which encompasses a range of disorders, and management can be carried out on a firm basis only if an accurate diagnosis is achieved (Scully and Porter, 1997). Clinical features such as a positive Nikolsky sign are not specific.
Therefore, in addition to a full history and examination, biopsy examination and appropriate histopathological and immunological investigations are frequently indicated. Biopsy of perilesional tissue, with histological and immunostaining examination, is essential to the diagnosis. Assay of serum antibody titers by indirect immunofluorescence (IIF) may also help guide prognostication and therapy. A recent critical evaluation of two ELISAs for the detection of antibodies to Dsg 1 and 3 comparing two substrates, normal human skin (HS) and monkey esophagus (MO), showed that, when PV serum was used, the sensitivity of IIF was 83% on HS and 90% on MO, and that this combination of substrates should not only increase the sensitivity of detecting pemphigus antibodies, but would also aid in the differentiation of PV from PF (Harman et al., 2000a). This strongly suggests that both substrates should be used in the diagnosis of PV, since patients with predominantly oral disease may have only Dsg3 antibodies, which are not always detectable in human skin.
Management
Oral lesions of pemphigus vulgaris may respond partially to topical or intralesional corticosteroids or other immunosuppressants. The treatment of DG consists of improving the oral hygiene, minimizing irritation of the lesions (Checchi et al., 1988), the use of specific therapies for the underlying disease where available, and often local immunosuppressive treatment (Lozada-Nur et al., 1991), but systemic immunosuppressive therapy, notably corticosteroids (Nisengard and Rogers, 1987), is almost inevitably required in PV.
In any event, in the absence of systemic treatment, oral lesions of PV are almost invariably followed by involvement of the skin or occasionally other epithelia such as the esophagus (Mignogna et al., 1997), when systemic immunosuppression will almost invariably be required.
Global immunosuppression is still largely used, though recently there have been attempts at more specific modulation of the autoimmune response which requires autoreactive helper T-cells that regulate immunoglobulin isotype switching. Systemic corticosteroids remain the mainstay of therapy of patients with oral lesions, transforming an invariably fatal disease into one whose mortality is now below 10% (Scully et al., 1999; Mignogna et al., 2000).
Current treatment, therefore, is largely based on systemic immunosuppression by systemic corticosteroids, with azathioprine, dapsone, methotrexate, cyclophosphamide, gold, and cyclosporin as adjuvants or alternatives, and this has significantly reduced the mortality (Mourellou et al., 1995; Bystryn and Steinman, 1996; Carson et al., 1996; Mobini et al., 1997b; Korman, 2000). The recognition that the severity of the disease is related to the proportions of Dsg3 and Dsg1 antibodies (Harman et al., 2000a) and to the titer of each (Harman et al., 2001b) suggests that sequential assays to monitor the specificity and titer of antibodies, along with the clinical features, may be useful in determining the degree of immunosuppression needed.
Hence, topical corticosteroids may suffice for a time if there are only localized oral lesions, with low-titer serum antibodies, but otherwise systemic immunosuppressants (e.g., prednisolone) are essential (Muller and Stanley, 1990; Lamey et al., 1992; Chrysomallis et al., 1994; Scully et al., 1999), and patients should be closely monitored (Table 4). Some use corticosteroids intravenously (Chrysomallis et al., 1995; Werth, 1996; Femiano et al., 2001) or use steroids with perhaps fewer adverse effects such as deflazocort (Mignogna et al., 2000). Once the disease is under clinical control, the dose of corticosteroid can be tapered (Rosenberg et al., 1976) or adjuncts added.
Thus, treatment is still largely with systemic corticosteroids, with steroid-sparing agents, and it remains to be seen whether newer therapies discussed below, such as mycophenolate mofetil or intravenous immunoglobulin therapy, prove in the longer term to offer significant advantages over the systemic corticosteroids.
Alternative treatments to corticosteroids
Adjuncts or alternatives to corticosteroids in the treatment of PV include several other immunosuppressive therapies. Chlorambucil (Shah et al., 2000), azathioprine (Roenigk and Deodhar, 1973), or cyclophosphamide (Lever and Schaumburg-Lever, 1977, 1984; Fellner et al., 1978; Piamphongsant, 1979; Ruocco, 1988; Pasricha et al., 1988, 1995) may be effective, the latter sometimes being effective when azathioprine is not (Ahmed and Hombal, 1987). Immunoablative high-dose cyclophosphamide without stem cell rescue has been successful in one patient (Hayag et al., 2000). Cyclosporin has proved effective in some hands (Balda and Rosenzweig, 1986; Cunliffe, 1987; Barthelemy et al., 1988; Mobini et al., 1997b) but not in others as an adjuvant to corticosteroids (Ioannides et al., 2000). However, methotrexate is not recommended (Carson et al., 1996). Adverse effects of these drugs are common (Scully and Cawson, 1998), but mycophenolate mofetil offers the hope of relatively safe immunosuppression with no nephrotoxicity or hepatotoxicity (Enk and Knop, 1997, 1999; Bredlich et al., 1999), and tacrolimus may have a place (Wu et al., 2002).
Other drugs
Other agents used with variable benefit include gold (Penneys et al., 1976; Salomon and Saurat, 1986), dapsone (Piamphongsant, 1979; Basset et al., 1987), etretinate (Orfanos and Bauer, 1983), prostaglandin E2 (Morita et al., 1995), and minocycline (Gaspar et al., 1996).
Plasmapheresis
Plasmapheresis (Cotterill et al., 1978; Blaszczyk et al., 1981; Swanson and Dahl, 1981; Roujeau et al., 1982; Bystryn, 1988; Roujeau, 1993; Turner et al., 2000) sometimes with cyclosporin (Ruocco, 1988) or cyclophosphamide (Kiel synchronization protocol) and extracorporeal photophoresis (Edelson, 1984) have also been reported to be of benefit.
Intravenous immunoglobulins
Intravenous immunoglobulins have proved successful and safe in steroid-resistant PV (Mobini et al., 1995; Bewley and Keefe, 1996; Bystryn and Steinman, 1996; Engineer et al., 2000; Sibaud et al., 2000).
Remission
The incidence of remissions in pemphigus is unclear, because these are usually reported at a single point in the evolution of the disease. Thus, it is uncertain whether treatment simply suppresses the manifestations of the disease and consequently must be continuously administered, or induces complete and long-lasting remissions that permit therapy to be discontinued. However, a recent long-term longitudinal study examined the induction of complete and long-lasting remissions (defined as “lesion-free with no systemic therapy for at least 6 months”) in 40 patients with PV treated conventionally and followed up for an average of 7.7 yrs, and showed that five (5%) of the patients died of the disease but that complete and long-lasting remissions were induced in 25%, 50%, and 75% of patients 2, 5, and 10 yrs, respectively, after diagnosis (Herbst and Bystryn, 2000). Most of the remaining patients were in partial remission or had mild disease controlled with a small dose of corticosteroids. The course of the disease followed different patterns, with some patients rapidly entering complete and long-lasting remissions, whereas others never entered into a complete remission. The induction of complete remission was related to the initial severity and extent of disease and to early response to treatment (Herbst and Bystryn, 2000).
It is thus possible, eventually, to induce complete and durable remissions in most patients, permitting systemic therapy to be safely discontinued without a flare in disease activity. The proportion of patients in whom this can be achieved increases steadily with time, and therapy can be discontinued in approximately 75% of patients after 10 years (Herbst and Bystryn, 2000).
Future Directions
The pathogenesis of PV is rapidly being unraveled, and the search for etiological factors may soon bear fruit. New, more effective, more specific, and safer treatments are emerging, over and above the recent newer immunosuppressive agents such as mycophenolate and tacrolimus, and include: proteinase inhibitors (Dobrev et al., 1996), chimeric molecules for specific recognition and elimination of the autoimmune B-cells (Proby et al., 2000), suggestions for targeting Dsg 3-specific T-cells for the eventual modulation of the T-cell-dependent production of pathogenic autoantibodies in PV (Hertl and Riechers, 1999), and suggestions for a novel avenue for the development of a non-steroidal treatment for PV using the anti-acantholytic activity of cholinergic agonists (Grando, 2000).
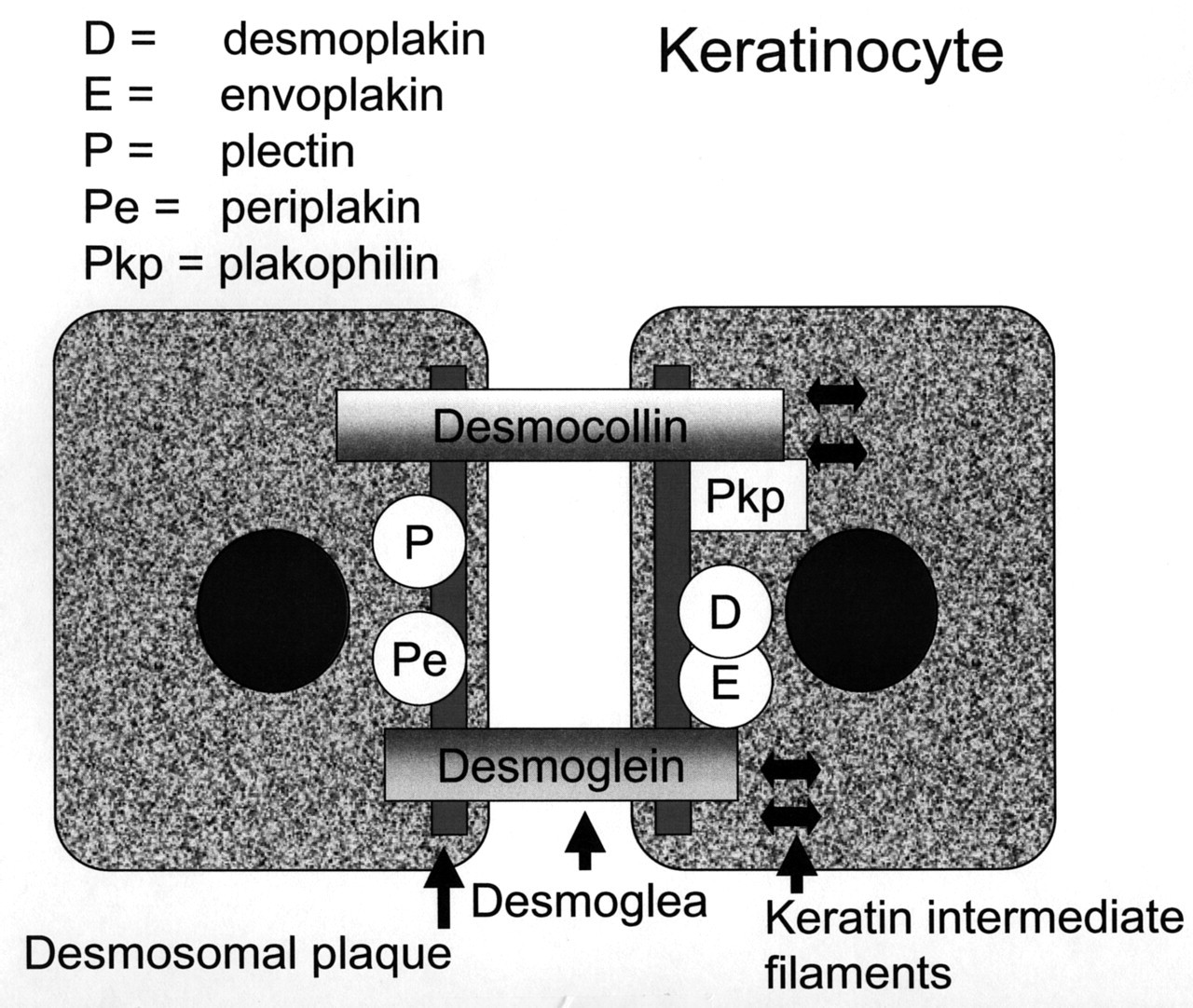
Epithelial structure.
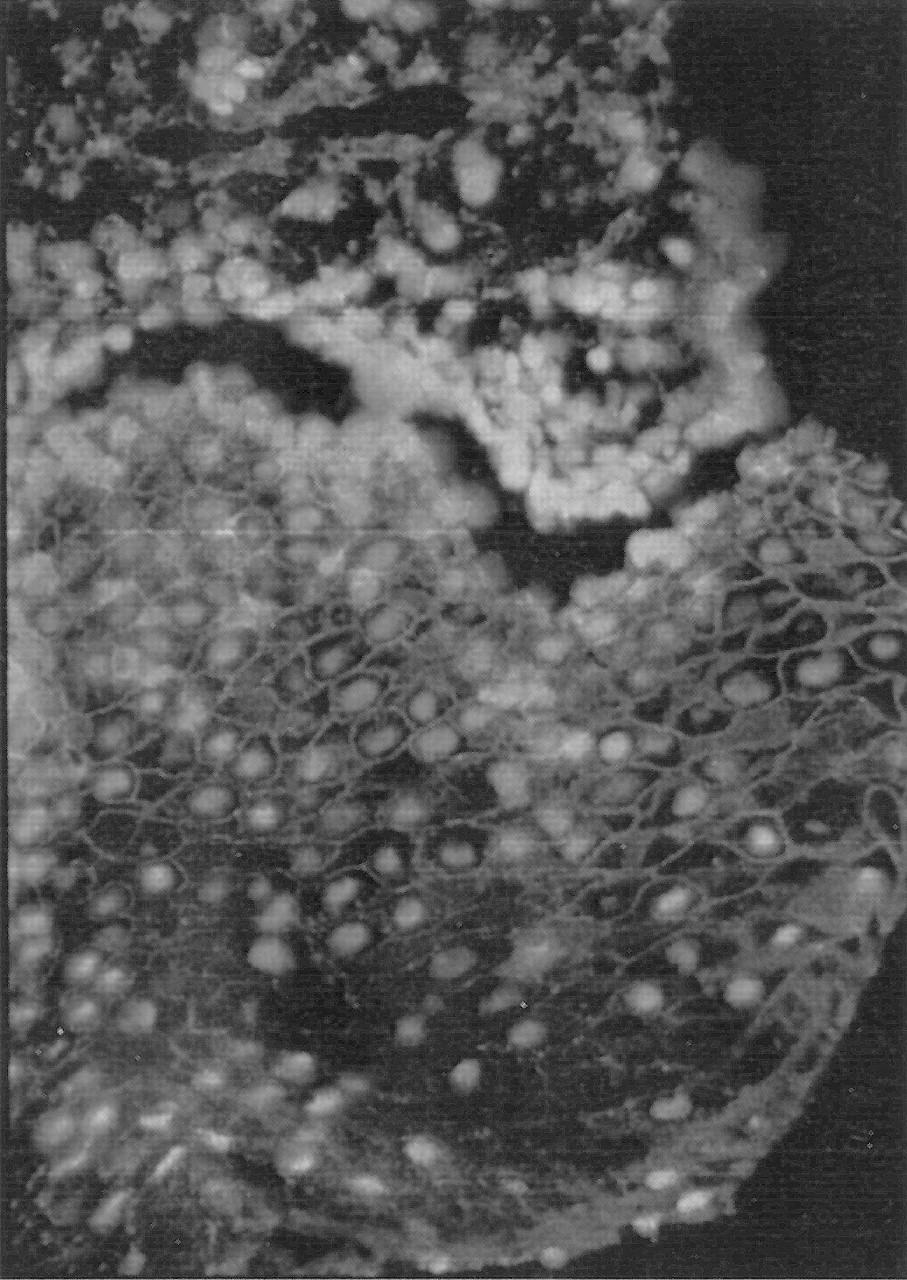
Immune deposits in PV.
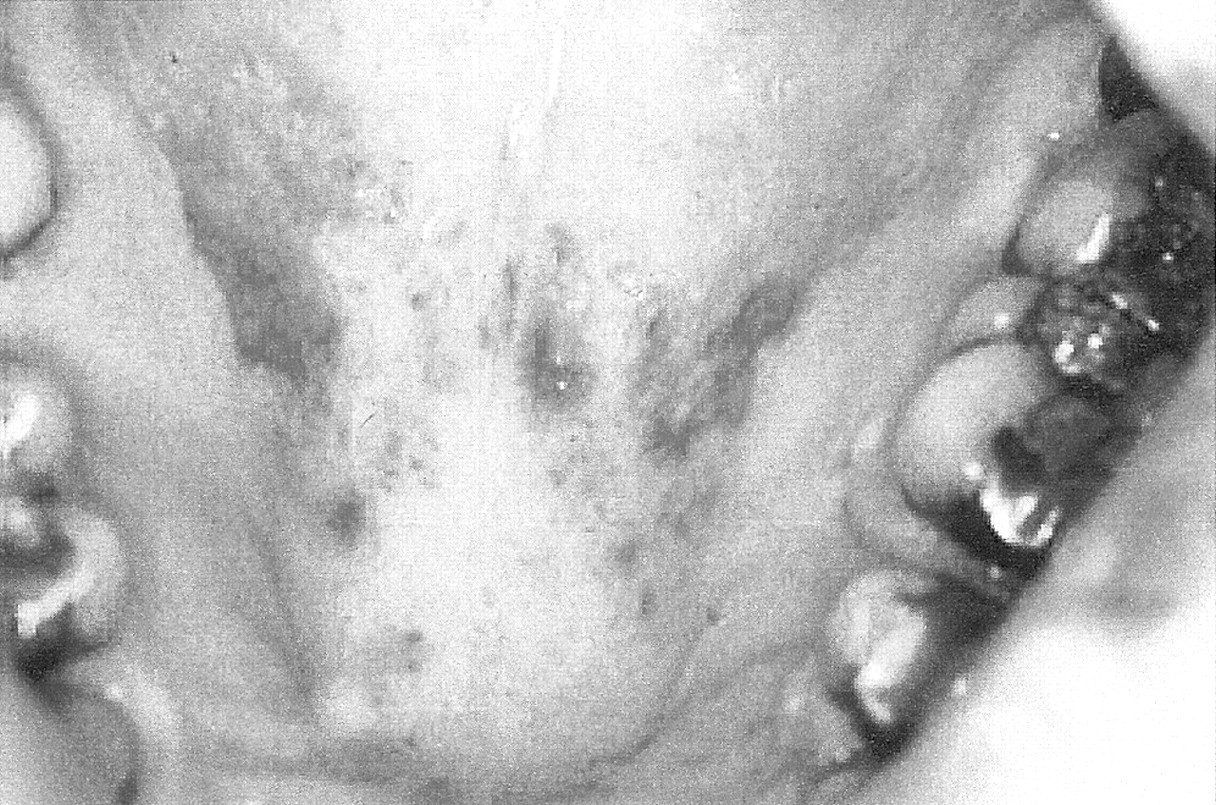
Oral lesions in PV.
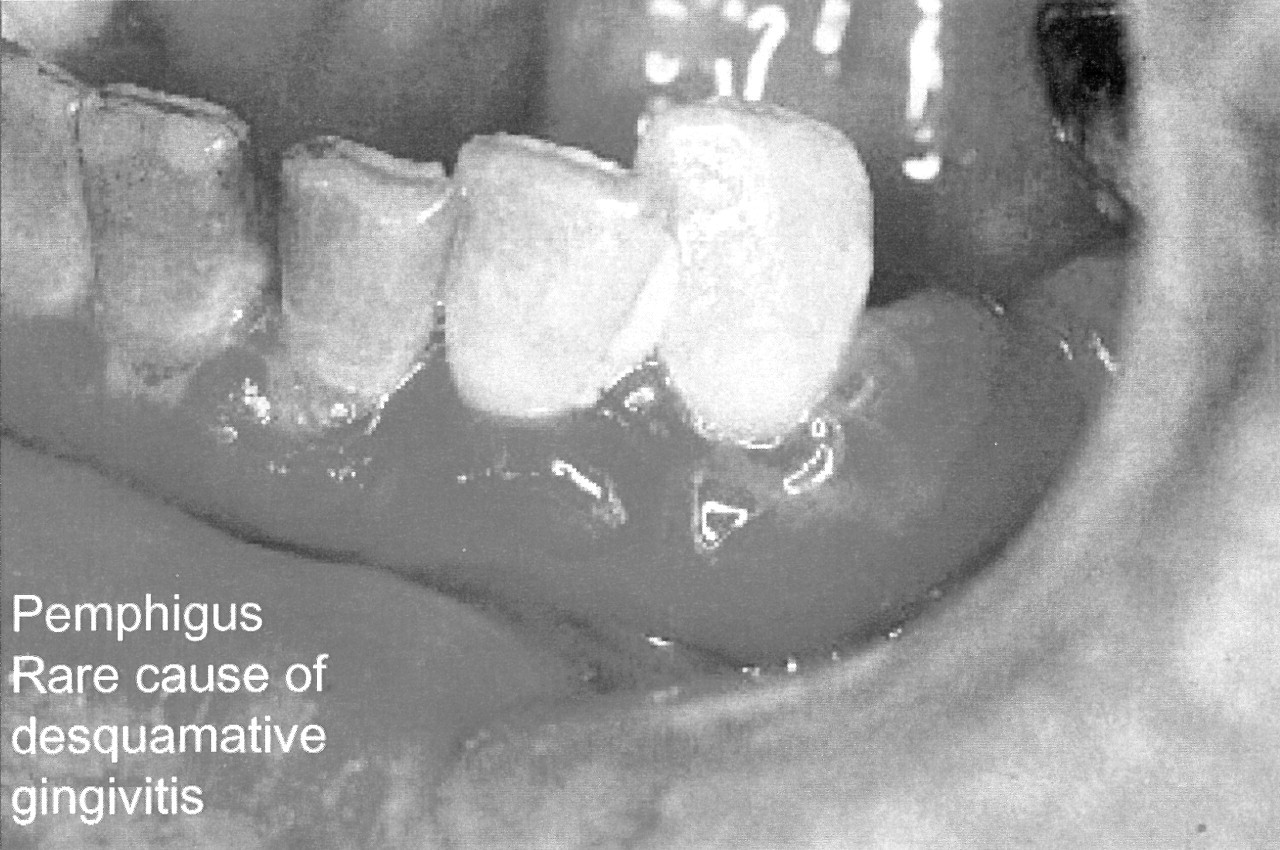
Oral lesions in PV.