
Abstract
The structurally related neuropeptides VIP and PACAP are released within the lymphoid organs following antigenic stimulation, and modulate the function of inflammatory cells through specific receptors. In activated macrophages, VIP and PACAP inhibit the production of pro-inflammatory agents (cytokines, chemokines, and nitric oxide), and stimulate the production of the anti-inflammatory cytokine IL-10. These events are mediated through the VIP/PACAP effects on de novo expression or nuclear translocation of several transcription factors, i.e., NFκB, CREB, c-Jun, JunB, and IRF-1. The in vivo administration of VIP/PACAP results in a similar pattern of cytokine and chemokine modulation, which presumably mediates the protective effect of VIP/PACAP in septic shock. In addition, VIP/PACAP reduce the expression of the co-stimulatory molecules B7.1/B7.2, and the subsequent stimulatory activity of macrophages for T-helper cells. In T-cells expressing specific VIP/PACAP receptors, VIP and PACAP inhibit the expression of FasL through effects on NFκB, NFAT, and Egr2/3. The reduction of FasL expression has several biological consequences: inhibition of antigen-induced cell death in CD4 T-cells, inhibition of the FasL-mediated cytotoxicity of CD8 and CD4 effectors against direct and bystander targets, and promotion of long-term memory Th2 cells, through a positive effect on the survival of Th2, but not Th1, effectors. The various biological effects of VIP and PACAP are discussed within the range of a general anti-inflammatory model.
(1) Introduction
The bi-directional interactions between the neuroendocrine and the immune systems are mediated by anatomical connections and mediators released and recognized by one or both systems. Both primary and secondary lymphoid organs and tissues possess an extensive peptidergic innervation close to the immune cells, with vasoactive intestinal peptide (VIP), neuropeptide Y (NPY), somatostatin (SOM), and galanin (Gal) as prevalent neuropeptides in the noradrenergic autonomic and cholinergic nerves, and substance P (SP), neurokinin A (NKA), and calcitonin gene-related peptide (CGRP) in the sensory innervation (Felten et al., 1987, 1992; Fink and Weihe, 1988; Bellinger et al., 1990; Nohr and Weihe, 1991; Weihe et al., 1991; Ichikawa et al., 1994). In addition, immune cells themselves express and release numerous neuropeptides (Lolait et al., 1986; Weinstock et al., 1990; Singaram et al., 1991; Przewlocki et al., 1992; Vollmar et al., 1992; Schwarz et al., 1994; Rajora et al., 1996; Brouxhon et al., 1998; Ryu et al., 2000). Some of these neuropeptides are released in functionally relevant amounts during the immune response, both in patients and in animal models (Nieber et al., 1992; Mosmann et al., 1993; Kaltreider et al., 1997).
The neuropeptides released within the lymphoid microenvironment and the corresponding neuropeptide receptors on immune cells represent the framework for neuropeptides mediating neuroimmune interactions. Although the list of immunomodulatory neuropeptides/neurotransmitters grows at a steady pace, here we review only the effects of VIP and PACAP, two of the best-studied immunoregulatory neuropeptides. The prominent role of VIP and PACAP in immunoregulation is reflected by the number of recent reviews related to this subject (Pozo et al., 2000; Ganea and Delgado, 2001a,b; Gomariz et al., 2001). Here, we focus on the most recent developments regarding the effects of VIP/PACAP on innate and adaptive immunity, particularly on macrophage activation and T-cell differentiation.
(2) VIP/PACAP Sources in the Immune Organs
VIP, discovered first (Said and Mutt, 1969), and PACAP, discovered 20 years later (Miyata et al., 1989), belong to a nine-member family that developed through gene and/or exon duplications, starting presumably with PACAP as the ancestral molecule (Sherwood et al., 2000). VIP and PACAP share the highest homology within the family, act on the same receptors, and share many biological activities. Although little is known about the presence of PACAP in the lymphoid organs, VIP has been studied in much more detail. VIP-ergic nerve fibers are found in most lymphoid organs, particularly in the respiratory and gastrointestinal tract, close to immune cells (Pearse et al., 1977; Dey et al., 1981; Bellinger et al., 1990; Kulkarni-Narla et al., 1999). The signals leading to neuronal VIP release during an inflammatory reaction have not yet been elucidated. A strong candidate is nitric oxide (NO), which functions as a particularly potent signal for VIP release from enteric ganglia (Grider and Jin, 1993; Allescher et al., 1996) and is produced at high levels in inflammation.
In addition, immune cells themselves express VIP at both mRNA and protein levels, and stimulators of immune cells such as LPS, cytokines, ConA, or anti-TCR antibodies act as inducers of VIP release (reviewed in Gomariz et al., 2001). We reported recently that VIP is expressed and released from Th2, but not Th1 cells, upon specific antigen stimulation, both in vivo and in vitro (Delgado and Ganea, 2001a). These results indicate that, similar to other neuropeptides, VIP and presumably PACAP are released in the lymphoid organs from two separate sources, the innervation and the immune cells. The challenge remains to identify the factors that control neuronal or immune release of VIP/PACAP during an inflammatory response and to characterize the molecular mechanisms involved.
(3) VIP/PACAP Receptors in Immune Cells
Three types of VIP/PACAP receptors have been cloned recently: VPAC1 and VPAC2 bind both VIP and PACAP with equal affinity, and activate primarily the adenylate cyclase pathway, and PAC1, the PACAP preferring receptor, binds PACAP with a 300- to 1000-fold higher affinity than VIP, and activates both adenylate cyclase and phospholipase C (Rawlings and Hezareh, 1996; Harmar et al., 1998). The characterization of VIP/PACAP receptors on various immune cell subpopulations is complicated by the modulation of some of these receptors upon cell activation and differentiation. For example, murine peritoneal macrophages, the murine macrophage cell line RAW 264.7, and the human monocytic cell line THP-1 express VPAC1 and PAC1 mRNA constitutively, and VPAC2 following LPS stimulation (Delgado et al., 1999a; Delgado and Ganea, 1999, 2001b). Expression of VPAC1, but not VPAC2, on resting human monocytes has also been reported (Lara-Marquez et al., 2001).
T-lymphocytes express VPAC1 and VPAC2, but not PAC1 (Delgado et al., 1996; Johnson et al., 1996; Xia et al., 1996; Kaltreider et al., 1997; Jiang et al., 1998; Busto et al., 2000). Induction of VPAC2 expression during T-cell activation and differentiation was suggested by several studies (Delgado et al., 1996; Metwali et al., 2000) and by the fact that several fully differentiated T-cell lines express only VPAC2 (Xin et al., 1997; Delgado and Ganea, 2000a). Recently, quantitative real-time RT-PCR studies indicated that VPAC1 is down-regulated and VPAC2 slightly up-regulated following stimulation of human blood T-cells (Lara-Marquez et al., 2001), and that VPAC2 is significantly up-regulated in murine CD4+ T-cells following activation with anti-CD3 and anti-CD28 Abs (Voice et al., 2001).
Whether the various receptors mediate different functions remains to be established. The inhibition of cytokine production by freshly activated T-cells appears to be equally mediated by VPAC1 and VPAC2 (Jiang et al., 1998). However, VPAC2, but not VPAC1, transduces human T-cell chemotaxis (Xia et al., 1996; Goetzl et al., 1998), and VPAC2 mediates the VIP-enhanced conversion of thymocytes into CD4+8- T-cells (Pankhaniya et al., 1998). In macrophages, most of the VIP/PACAP effects appear to be mediated by VPAC1, with the exception of the IL-6 inhibition, which is mediated by PAC1 (reviewed in Delgado et al., 1999b).
(4) Effects of VIP/PACAP on Innate Immunity
Macrophages play a crucial role in the fight against pathogens, by contributing to both innate and adaptive immunity. Phagocytosis of pathogens is a characteristic of macrophages, leading to their activation in terms of cytokine production and antigen presentation, and to the reduction of the pathogen load. Different studies reported opposite effects of VIP/PACAP on macrophage phagocytosis, adherence, migration, and superoxide production (reviewed in Ganea and Delgado, 2001b; Gomariz et al., 2001). The most probable explanation is that macrophages in different stages of activation express different VIP/PACAP receptors. Indeed, the transduction pathways involved in the stimulatory vs. inhibitory effects of VIP/PACAP indicate use of different receptors (Ganea and Delgado, 2001b).
(A) Effects of VIP/PACAP on the production of macrophage-derived cytokines: transduction pathways and transcription factors
Macrophages initiate the inflammatory response through the secretion of inflammatory cytokines and the production of reactive oxygen and nitrogen intermediates. Microbial products such as LPS induce macrophages to secrete several pro-inflammatory products such as tumor necrosis factor (TNFα), interleukin-12 (IL-12), interleukin 1 (IL-1), interleukin 6 (IL-6), and nitric oxide (NO), followed by the secretion of the anti-inflammatory cytokines interleukin-10 (IL-10) and transforming growth factor beta (TGFβ) (Laskin and Pendino, 1995). Although beneficial in host defense, a sustained production of pro-inflammatory agents has serious pathological consequences (Van Snick, 1990; Vassalli, 1992; Evans, 1995). The balance between pro- and anti-inflammatory factors plays an essential role in the successful control of inflammation. Regulatory molecules called “macrophage deactivating factors” have received considerable interest lately. In addition to the anti-inflammatory cytokines IL-10, IL-11, TGFβ, and IL-13 (Fiorentino et al., 1991; Trepicchio et al., 1996; Muchamuel et al., 1997; Tsunawaki et al., 1998), neuropeptides such as VIP, PACAP, CGRP, somatostatin, and α melanocyte-stimulating hormone (αMSH) also function as macrophage-deactivating factors (reviewed in Ganea and Delgado, 2001a). VIP/PACAP inhibit the in vitro and in vivo production of the pro-inflammatory cytokines TNFα, IL-6, IL-12, and of nitric oxide, and stimulate the production of the anti-inflammatory cytokine IL-10 (Dewit et al., 1998; Martinez et al., 1998; Xin and Sriram, 1998; Delgado et al., 1999a,c d e) (Fig. 1). In addition, VIP/PACAP exert a protective effect in vivo in models of septic shock. The protective effect correlates with the inhibition of endogenous pro-inflammatory cytokines and is mediated through the VPAC1 receptor (Delgado et al., 1999f, 2000).
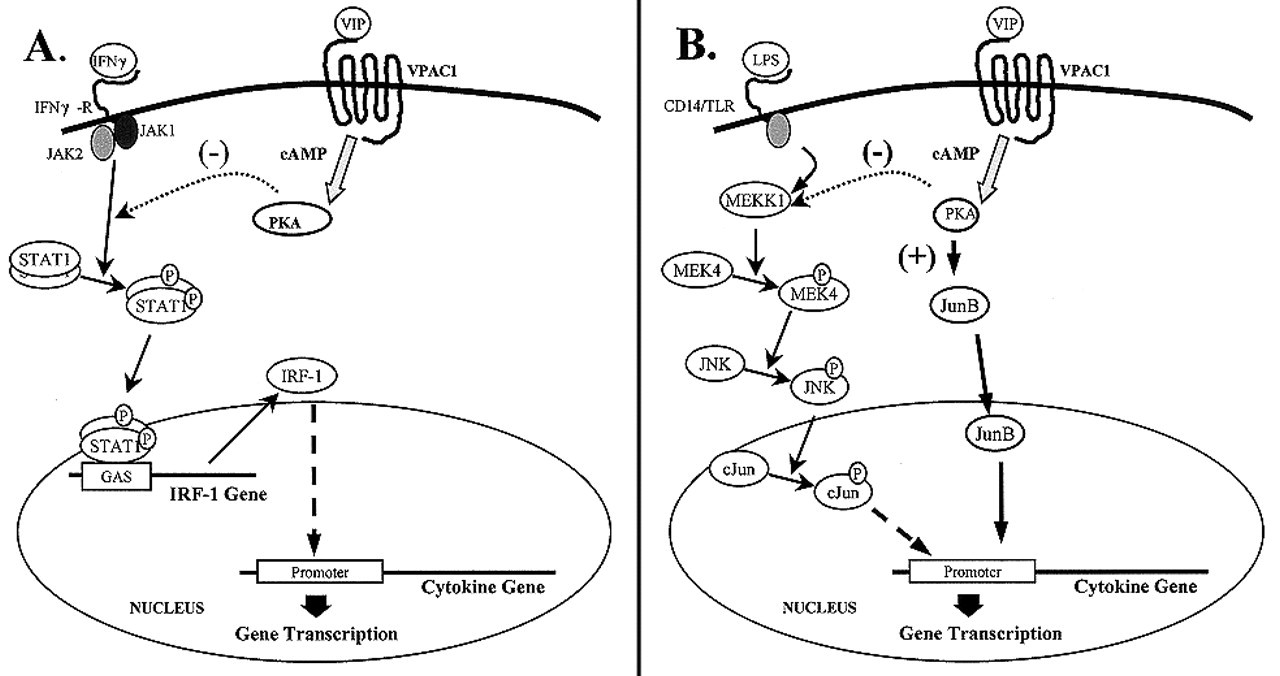
As expected from the involvement of VPAC1, the cAMP-dependent pathway plays a major role in the inhibition of TNFα, IL-12, and iNOS, and in the stimulation of IL-10 (Delgado et al., 1998, 1999d,e; Delgado and Ganea, 1999). However, except for IL-10, two pathways, a cAMP-dependent and a cAMP-independent pathway, appear to be involved in the inhibitory effects of VIP/PACAP. The effects of VIP/PACAP on both macrophage-derived cytokines and NO are exerted at the transcriptional level through the regulation of several transcription factors. In the case of TNFα, iNOS, and IL-12p40 inhibition, the cAMP-independent pathway leads to the reduction in NFκB binding (Delgado et al., 1998, 1999d; Delgado and Ganea, 1999). In addition, the cAMP-dependent pathway mediates changes in the CRE-binding complex from highc-Jun/lowCREB to lowc-Jun/highCREB for the TNFα promoter (Delgado et al., 1998), inhibition of IRF-1 binding for the iNOS and IL-12p40 promoters (Delgado et al., 1999d; Delgado and Ganea, 1999), and changes in the composition of the AP-1 complexes from c-Jun/c-Fos to JunB/c-Fos with subsequent reduction in AP-1 binding to the TNFα promoter (Delgado and Ganea, 2000b). The VIP/PACAP increase in IL-10 gene transcription is mediated entirely through an increase in cAMP-dependent CREB binding (Delgado et al., 1999e).
Some of the detailed pathways leading from VPAC1 to gene transactivation have been elucidated (Figs. 2A-2D). Reduction in IRF-1 binding and synthesis is mediated through the inhibition of IFNγ-induced Jak1/2-STAT1 phosphorylation (Fig. 2A); although the step(s) between cAMP induction and inhibition of Jak1/2 phosphorylation are not known, we eliminated the involvement of SOCS1 and 3 (Delgado and Ganea, 2000c). Changes in the composition of the AP-1 complexes from c-Jun/c-Fos to JunB/c-Fos, with a subsequent decrease in AP-1 binding, are mediated through the inhibition of the MEKK1/MEK4/JNK pathway, which leads to a reduction in c-Jun phosphorylation and a direct, positive effect on JunB synthesis (Delgado and Ganea, 2000b) (Fig. 2B). The VIP/PACAP decrease in NFκB binding which affects numerous genes involved in the inflammatory response (cytokines, chemokines, iNOS) is due to a more complex mechanism. The cAMP-independent pathway results in the inhibition of IκB phosphorylation, and its subsequent stabilization. Due to the prolonged presence of this inhibitor, p65 (an essential component of the NFκB transactivating complex) is retained in the cytoplasm. The VIP/PACAP effect on IkB phosphorylation is mediated through the inhibition of IKK, the IkB-specific kinase (Delgado and Ganea, 2001b) (Fig. 2C). For maximal transactivation, NFκB is further complexed to co-activators such as CBP (the CREB-binding protein) and phosphorylated TBP (the TATA-box binding protein) (Yang et al., 1996). VIP/PACAP affect both these proteins in a cAMP-dependent manner. First, through the increase in CREB phosphorylation, CBP is sequestered by nuclear phosphorylated CREB, instead of binding to p65 (Delgado and Ganea, 2001b) (Fig. 2C). Second, through the inhibition of the MEKK1/MEK3-6/p38 pathway, TBP phosphorylation is inhibited, leading to a decrease in TBP binding to both DNA and p65 (Delgado and Ganea, 2001b) (Fig. 2C). Finally, increases in CREB phosphorylation lead to increased CREB binding to the IL-10 promoter and activation of gene transcription (Delgado et al., 1999e) (Fig. 2D).
(B) Effects of VIP/PACAP on the production of macrophage-derived chemokines
The majority of immune cells are circulating, and directional movement is needed for homing to the right lymphoid compartment and to the site of pathogen invasion. This directional movement is controlled by chemokines released by a variety of cells and chemokine receptors expressed on the target immune cells. Although several studies have addressed the effects of VIP on lymphocyte adhesion and traffic (Moore et al., 1988; Bondesson et al., 1991; Johnston et al., 1994; Xia et al., 1996; Schratzberger et al., 1998), the reports are in little agreement beyond the fact that indeed VIP affects lymphocyte traffic. This is probably due to the fact that both chemokine production and expression of adhesion molecules and chemokine receptors depend on the cellular activation and differentiation stage, and that neuropeptides might affect all these processes. At present, there is only one report regarding the effects of VIP/PACAP on immune chemokine production, but no information as to their effects on chemokine receptors. We reported that VIP and PACAP inhibit the expression of two CXC chemokines (MIP-2 and IL-8) and of four CC chemokines (MIP-1α, MIP-1β, MCP-1, and Rantes), in vivo and in vitro. The inhibition is mediated by VPAC1 and correlates with a reduction in NFκB binding and transactivating activity (Delgado and Ganea, 2001c). The two CXC chemokines MIP-2 and IL-8 function as chemoattractants for neutrophils, whereas the CC chemokines attract monocytes/macrophages and T-cells. Accordingly, i.p. administration of VIP or PACAP in a model of acute peritonitis led to a significant reduction in the recruitment of neutrophils (from 12 to 24 hrs), and of macrophages and lymphocytes (from 24 to 48 hrs) in the peritoneal cavity (Delgado and Ganea, 2001c).
The possibility that neuropeptides, particularly VIP and PACAP, might modulate the expression of not only chemokines but also chemokine receptors opens new territories in neuroimmunomodulation, since differences in chemokine receptors impose the different migratory patterns of immature vs. mature dendritic cells, and of Th1 vs. Th2 cells (Sallusto et al., 1998; D'Amico et al., 2000; Sozzani et al., 2000; Zlotnik and Yoshie, 2000). Therefore, neuropeptides might be active participants in the maturation and function of dendritic cells, and in the differential development of Th1 and/or Th2 effector cells.
(C) Effects of VIP/PACAP on the expression of the co-stimulatory B7 molecules on resting and activated macrophages
In addition to innate immunity, macrophages also participate in adaptive immunity as antigen-presenting cells. To activate T-cells, macrophages have to provide a second, co-stimulatory signal, in addition to the MHC class II/antigen complex recognized by the specific T-cell receptor. Among the co-stimulatory molecules, B7.1 and B7.2 play major roles. In macrophages, B7.1 and B7.2 are expressed only following activation, with B7.2 being induced earlier and at higher levels than B7.1 (Lenschow et al., 1996).
VIP and PACAP are among the endogenous factors that regulate B7 expression in macrophages. Interestingly, the two neuropeptides affect B7 expression in resting and activated macrophages in an opposite manner. In resting macrophages, VIP/PACAP up-regulate B7.2, but not B7.1, expression at mRNA and protein levels, both in vivo and in vitro. In contrast, in LPS/IFNγ-activated macrophages, VIP/PACAP down-regulate both B7.1 and B7.2 expression (Delgado et al., 1999g). The effects of VIP/PACAP on B7 expression correlate with effects on the stimulatory activity for T-cells (Delgado et al., 1999g). The inhibition of B7.1/B7.2 expression in activated macrophages is in perfect agreement with the accepted role of VIP and PACAP as endogenous anti-inflammatory agents. Moreover, since VIP/PACAP affect B7 but not MHC class II expression (Delgado et al., 1999g), the neuropeptides could actually contribute to peripheral tolerance by inducing T-cell anergy. In contrast, the up-regulation of B7.2 in unstimulated macrophages might represent one of the mechanisms by which VIP/PACAP support Th2 differentiation, which is discussed below.
(5) Effects of VIP/PACAP on T-cell Function, Survival, and Differentiation
In agreement with the anti-inflammatory function of VIP/PACAP, several authors reported inhibitory effects on T-cell proliferation and IL-2 production (reviewed in Gomariz et al., 2001). However, the effects of VIP and PACAP on T-cell survival and differentiation are actually more complex, suggesting a more subtle immunomodulatory role for these neuropeptides.
(A) VIP/PACAP effects on antigen-induced apoptosis in T-cells
Following initial TCR engagement, antigen-specific CD4 T-cells enter a proliferative stage and differentiate into clones of antigen-specific effector T-cells. The termination of an immune response requires the elimination of the vast majority of antigen-specific T-cells through apoptosis. Although during initial activation T-cells are apoptosis-resistant, cycling T-cells become highly susceptible to apoptosis. Two different types of apoptosis, passive and active, eliminate activated T-cells. In the absence of further antigenic stimulation, the cycling T-cells undergo passive apoptosis, due to withdrawal of growth factors. However, if the antigen persists, the cycling cells are eliminated through active apoptosis, or antigen-induced cell death (AICD), upon TCR re-engagement (reviewed in Lenardo et al., 1999).
Although studies in the CNS showed that VIP and PACAP act as neuronal survival factors in various injury conditions (Uchida et al., 1996>; Gressens, 1999; Reglodi et al., 2000; Gozes and Brenneman, 2000), the expectation in the immune system was that VIP/PACAP would promote lymphocyte apoptosis, based on their general anti-inflammatory role. However, contrary to this expectation, VIP and PACAP protect activated CD4 T-cells against AICD both in vitro and in vivo (Delgado and Ganea, 2000a). CD4 T-cells undergo AICD primarily through Fas/Fas ligand (FasL) interactions (Alderson et al., 1995; Ju et al., 1995). Fas is expressed constitutively on CD4 T-cells and up-regulated following activation, whereas FasL expression is induced only following TCR re-stimulation (Suda et al., 1995). The VIP/PACAP protection against AICD is actually the result of an inhibitory effect on FasL expression in the re-stimulated CD4 T-cells (Delgado and Ganea, 2000a). The inhibition of FasL expression is mediated through effects on several transcription factors, i.e., NFκB, NF-ATp, and Egr2,3 (Delgado and Ganea, 2001d). Similar to their effect in macrophages, VIP/PACAP stabilize IkB and prevent p65 nuclear translocation in T-cells. In addition, VIP/PACAP inhibit NF-ATp nuclear translocation in a calcineurin-independent manner, and prevent the expression of Egr2 and 3, presumably indirectly, through the inhibition of NF-ATp nuclear translocation. In contrast to macrophages, where the effects on NFκB are mostly cAMP-independent, in T-cells cAMP functions as the secondary messenger in affecting all three transcription factors (NFκB, NFAT, Egr2/3) (Delgado and Ganea, 2001d).
The different subsets of effector Th cells play different roles in the immune response, with Th1 being primarily involved in cell-mediated immunity, and Th2 in humoral immunity. The two subsets appear to differ also in terms of susceptibility to AICD, with Th2 cells being more resistant to apoptosis (Varadhachary et al., 1997; Zhang et al., 1997). Whether VIP and PACAP affect AICD preferentially in one of the Th subsets remains to be determined. Recent results from our laboratory suggest that indeed VIP/PACAP protect Th2 cells preferentially, and that this protection correlates with the inhibition of FasL expression (Delgado and Ganea, submitted).
(B) VIP/PACAP inhibit Fas L-mediated cytotoxicity against direct and bystander targets
FasL/Fas interactions are involved not only in CD4 T apoptosis, but also in cytotoxicity against Fas-bearing targets. Cytotoxic CD8 T-cells (CTL) kill targets through a calcium-dependent, perforin/granzyme-mediated, and through a calcium-independent, FasL-mediated mechanism (Berke, 1995). VIP and PACAP do not affect the perforin/granzyme-mediated cytotoxicity, but inhibit drastically the FasL-mediated lysis of both allogeneic and syngeneic Fas-bearing targets. VIP/PACAP inhibit the FasL-mediated cytotoxicity by preventing FasL expression on the cytotoxic T-cells both in vivo and in vitro (Delgado and Ganea, 2000d).
CD4 T effector cells also express cytotoxic activity. However, in contrast to the CD8 CTLs, the CD4 lytic effectors kill targets only through FasL/Fas-mediated interactions (Ju et al., 1994; Stalder et al., 1994). Antigen-presenting cells (APC) activate CD4 T-cells in a MHC II-restricted manner, leading to the up-regulation of FasL, which then enables the CD4 T-cells to lyse both cognate APCs (direct targets) and neighboring Fas-bearing cells (bystander targets) in an antigen-independent and MHC-nonrestricted manner (Wang et al., 1996; Smyth, 1997). VIP and PACAP inhibit FasL expression in allogeneic and antigen-specific CD4 effectors generated in vivo, and their cytotoxicity for both direct and bystander targets (Delgado and Ganea, 2001e). The elimination of activated APCs removes potentially harmful cells after the completion of an immune response, and therefore represents a required mechanism for the maintenance of immune homeostasis. However, bystander killing results in collateral damage, particularly relevant for tissues with limited MHC II expression, such as the brain. In experimental allergic encephalomyelitis (EAE), myelin-specific CD4 T effectors contribute to the pathology, through lysis of antigen-non-specific targets (Thilenius et al., 1999). Similar mechanisms may be responsible for tissue damage in other autoimmune diseases as well (De Maria and Testi, 1998). In this respect, endogenous agents such as VIP and PACAP, capable of down-regulating FasL expression on T-cells, may be important in the control of FasL/Fas-mediated lysis of innocent bystanders, particularly in immune privileged organs such as the brain and the anterior chamber of the eye, where these neuropeptides are abundant.
(C) Effects of VIP/PACAP on CD4 T-cell differentiation
Following antigenic stimulation, CD4 T-cells differentiate into Th1 and Th2 effector cells, characterized by specific cytokine profiles and functions. Determining factors for the differentiation include the nature of the APC, the nature and amount of antigen, and the genetic background of the host, with the cytokine microenvironment as the dominant factor (reviewed in O'Garra, 1998). Among cytokines, IL-12 and IL-4 appear to be the determinant factors for Th1 and Th2 differentiation, respectively. The possible role of neuropeptides in Th1/Th2 differentiation is just beginning to be investigated.
VIP and PACAP induce Th2 responses in vivo and in vitro (Delgado et al., 1999h). Macrophages treated in vitro with VIP or PACAP gain the ability to induce Th2-type cytokines (IL-4 and IL-5) and inhibit Th1-type cytokines (IFNγ, IL-2) in Ag-primed CD4 T-cells. In vivo administration of VIP or PACAP in Ag-immunized mice results in a decreased number of IFNγ-secreting cells and an increased number of IL-4-secreting cells (Delgado et al., 1999h). The initial observation of a Th2 bias by VIP/PACAP in normal mice has been confirmed and extended by two elegant studies with VPAC2 transgenic and knockout mice (Voice et al., 2001, and Goetzl et al., 2001, respectively). The two models present opposite phenotypes. The transgenic mice constitutively expressing VPAC2 in CD4+ T-cells produce significantly more Th2 than Th1 cytokines, have high levels of IgE and eosinophils, and develop cutaneous allergic reactions. In contrast, in VPAC2-deficient mice, T-cell activation results in the production of significantly more Th1 than Th2 cytokines, leading to enhanced delayed-type hypersensitivity and depressed cutaneous anaphylactic reaction.
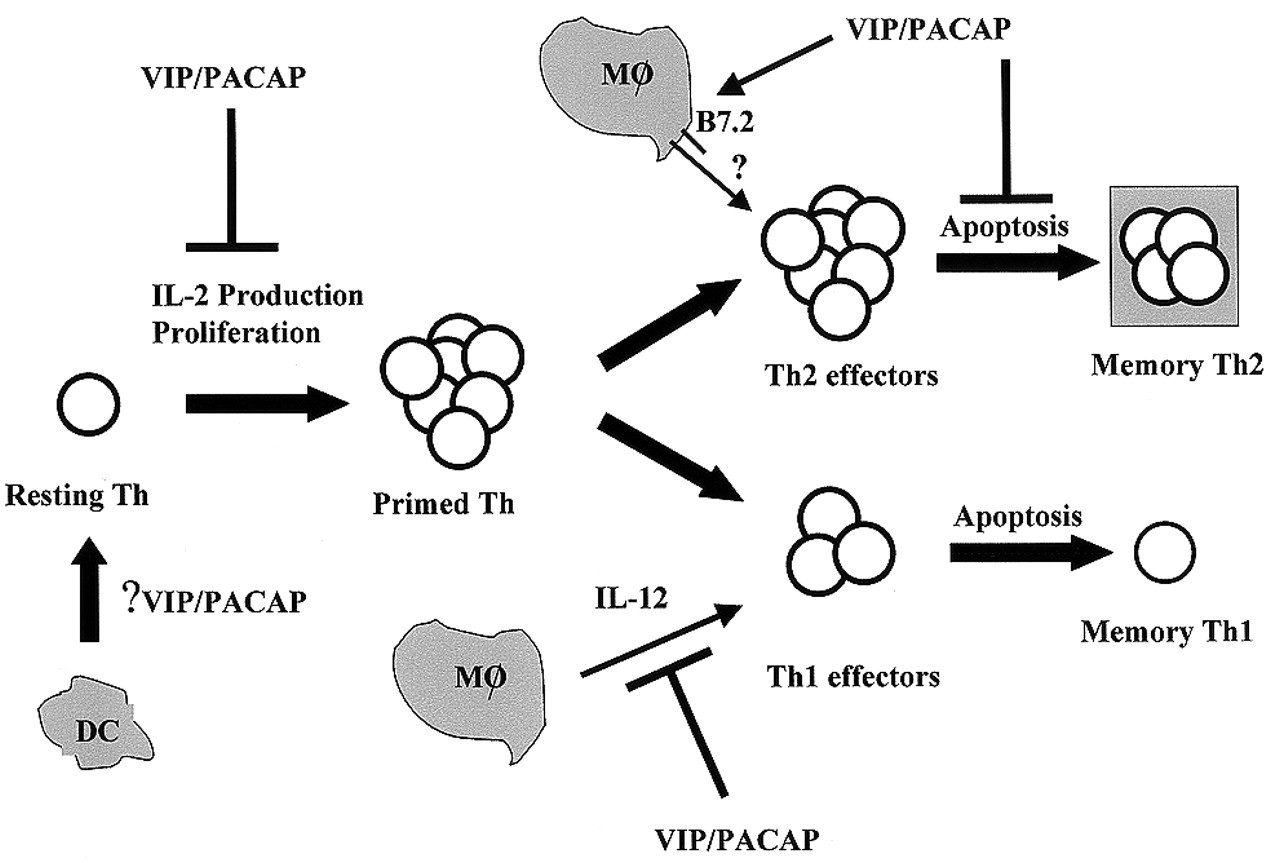
There are several possible non-excluding mechanisms for the VIP/PACAP bias toward Th2 (Fig. 3). Inhibition of macrophage IL-12 production by VIP and PACAP is one possibility. Since IL-4 dominates IL-12, driving naïve CD4 T-cells toward the Th2 phenotype (O'Garra, 1998), a reduction in IL-12 by VIP/PACAP, even in the absence of an effect on IL-4, will result in Th2 differentiation. A second possibility is up-regulation of B7.2 expression on macrophages by VIP/PACAP. Although the role of B7.1 and B7.2 in selectively activating Th1 or Th2-type responses is controversial, several disease models suggest a role for B7.2 in promoting Th2 differentiation (Kuchroo et al., 1995; Keany-Myers et al., 1998; Larche et al., 1998). Finally, a very recent study from our laboratory indicates that VIP and PACAP support Th2, but not Th1, proliferation in vitro, and promote the generation of memory Th2 cells in vivo. These effects correlate with a reduction in FasL expression and inhibition of apoptosis in Th2, but not Th1, cells (Delgado and Ganea, submitted).
The fact that VIP and PACAP promote Th2 differentiation and/or survival appears at first glance contrary to the accepted anti-inflammatory role of these neuropeptides. However, since the final outcome is the predominant development of a Th2 response, with concomitant inhibition of Th1 differentiation, and since inflammatory reactions typical for cell-mediated immunity are associated with Th1 responses, the VIP/PACAP inhibition of Th1 development may represent an additional mechanism for their general anti-inflammatory activity. The fact that VIP and PACAP promote the generation and long-term survival of Th2 cells is particularly relevant in view of the concept that immune privilege in organs such as the brain and eye is an active process of immune deviation mediated by regulatory T-cells generated in the presence of Th2-derived cytokines (Harling-Berg et al., 1999; Takahashi et al., 1999). It is tempting to speculate that neuropeptides such as VIP and PACAP, which are abundant in the immune privileged sites, play an essential role in maintaining immune deviation. This is supported by the fact that immune privilege in the eye strictly depends on the presence of intact corneal nerves, being lost upon nerve severing and regained once the nerves regrow (Streilein et al., 2000).
(6) Conclusions
VIP, and the structurally related neuropeptide PACAP, released in the lymphoid organs by the innervation and by activated immune cells, modulate the function of inflammatory cells through specific receptors, affecting both innate and adaptive immunity. One of the major targets for VIP/PACAP activity is the stimulated macrophage. Through the release of cytotoxic molecules, including cytokines, oxygen radicals, and nitric oxide, macrophages decrease the pathogen load. The subsequent release of pro-inflammatory cytokines, the processing and presentation of the antigen, and the up-regulation of co-stimulatory molecules position the macrophage as an important participant in the induction of adaptive immunity. Responding to stimulatory and co-stimulatory signals delivered by APCs (including dendritic cells, B-cells, and macrophages), CD4 T-cells proliferate and differentiate into effector Th1 and Th2 cells. At the conclusion of an immune response, both activated APCs and T-cells have to be deactivated and/or eliminated. In the absence of the deactivation/elimination of stimulated immune cells, excessive tissue and organ damage leads to pathological conditions, and even death. Several endogenous factors, particularly anti-inflammatory cytokines, contribute to the down-regulation of the immune response. Neuropeptides, such as VIP and PACAP, can be added to the list of endogenous anti-inflammatory molecules. VIP and PACAP exert their anti-inflammatory function in several ways: (1) by direct inhibition of pro-inflammatory cytokine production (TNFα, IL-6, IL-12) by activated macrophages; (2) by up-regulation of IL-10 production (a potent anti-inflammatory cytokine); (3) by inhibition of B7.1/B7.2 expression in activated macrophages and subsequent inhibition of their stimulatory activity for antigen-specific T-cells; (4) by inhibition of IL-2 production and T-cell proliferation; (5) by inhibition of Th1 responses (reduction in both the amounts of Th1 cytokines and the number of cytokine-producing Th1 cells); and (6) by inhibition of FasL/Fas-mediated cytotoxicity of CD8 and CD4 T-cells against direct and bystander targets. In contrast to these well-defined anti-inflammatory functions, VIP and PACAP support the generation and long-term survival of Th2 cells, representing the first neuropeptides with a possible role in the generation of memory Th2 cells. In immune privileged sites such as the brain and the anterior chamber of the eye, this might be one of the most important physiological functions of VIP and PACAP, since the state of immune deviation depends on regulatory cells whose development requires Th2 cytokines.
Effects of VIP and PACAP on activated macrophages. VIP or PACAP released in the vicinity of activated macrophages bind to specific receptors (VPAC1, VPAC2, PAC1) and inhibit expression and production of pro-inflammatory agents (TNFα, IL-6, nitric oxide [NO], chemokines [CK], and IL-12) and, indirectly, through IL-12, VIP, and PACAP inhibit IFNγ production from antigen-stimulated Th cells. In contrast, VIP and PACAP up-regulate the production of IL-10, an anti-inflammatory cytokine. VIP and PACAP also inhibit expression of the co-stimulatory molecules B7.1 and B7.2, and the subsequent induction of T-cell proliferation. The in vivo inhibitory effects on pro-inflammatory cytokines and the up-regulation of the anti-inflammatory cytokine IL-10 correlate with the protective effects of VIP and PACAP against septic shock (Delgado et al., 1999f) and in experimental arthritis (Delgado et al., 2001). Macrophage-derived molecules inhibited by VIP/PACAP are boxed in shaded areas, whereas those up-regulated by VIP/PACAP are boxed in clear areas. Transduction pathways and transcription factors involved in the effects of VIP and PACAP on activated macrophages. Effects of VIP/PACAP on Th cell differentiation. Although recent reports indicate that VIP affects dendritic cell (DC) maturation and migration (Delneste et al., 1999; Dunzendorfer et al., 2001), the effects of VIP and PACAP on dendritic cell (DC)–resting Th cell interactions remain to be investigated. VIP/PACAP inhibit IL-2 production and T-cell proliferation following initial exposure to the antigen. VIP/PACAP affect differentiation into effector Th cells, inhibiting Th1 and promoting Th2 differentiation. This effect is exerted through at least two separate pathways: (a) inhibition of IL-12 production (a required Th1 differentiation factor); and (b) posibly, through the up-regulation of macrophage B7.2, but not B7.1, by VIP/PACAP. Generation of memory Th cells requires survival of a small number of activated effectors following apoptosis, which eliminates most effectors at the end of the immune response. VIP/PACAP promote the survival of Th2, but not Th1, effectors, and the generation of long-term memory Th2 cells.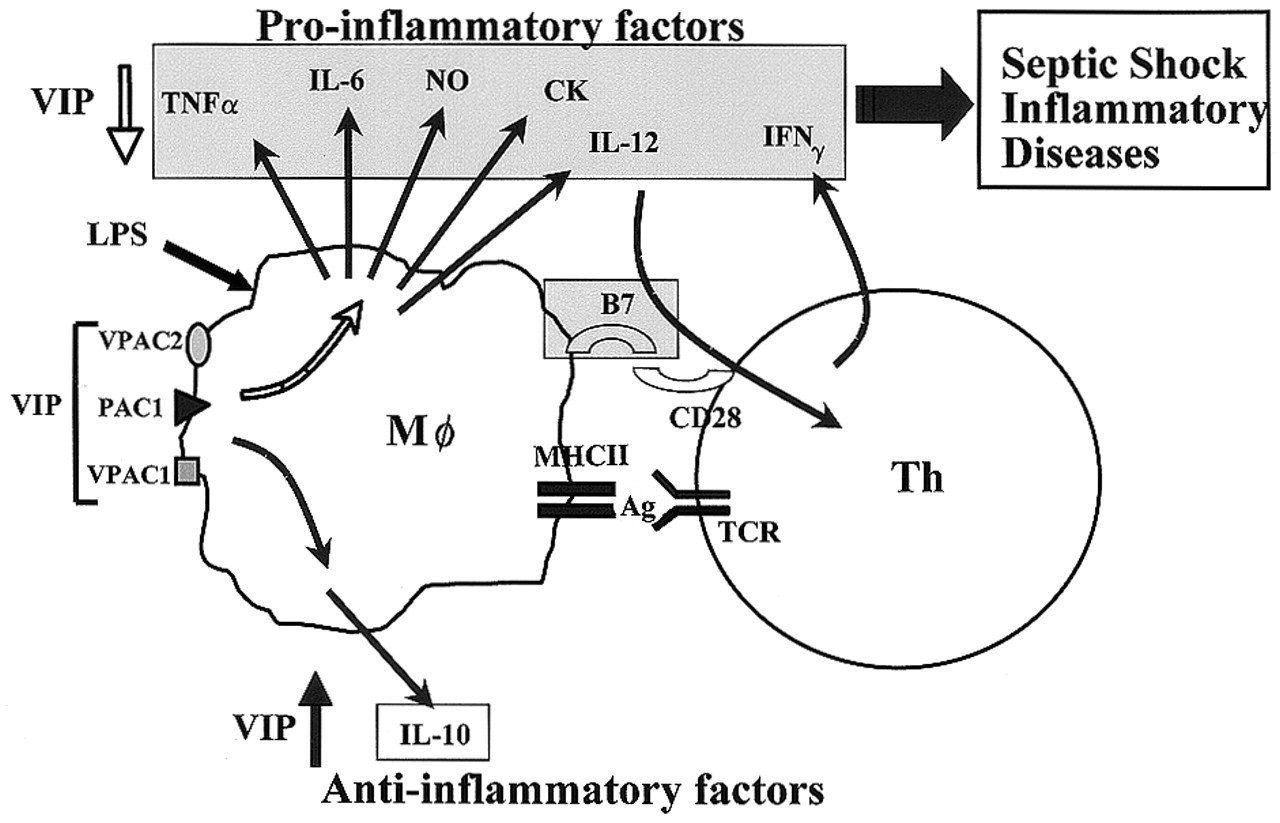
Footnotes
Acknowledgements
This work was supported by grants PHS AI41786-03 (DG) and AI47325-01(DG), by grant PM98-0081 (MD), and by a post-doctoral fellowship from the Spanish Department of Education and Science (MD).