
Abstract
Melanoma development not only involves genetic and epigenetic changes that take place within the cell, but also involves processes determined collectively by micro-environmental factors, including cell-cell interactions and communications. During the transition from normal cells to benign and malignant lesions, and subsequently to metastatic cancer, stepwise changes in intercellular communications provide tumor cells with the ability to overcome cell-cell adhesion and micro-environmental controls from the host and to invade surrounding tissues and disperse to distant locations. Cadherins are major cell–cell adhesion molecules involved in the development and maintenance of skin. E-cadherin expressed in normal melanocytes mediates growth and invasion control by keratinocytes. Progressive loss of E-cadherin and gain of N-cadherin during melanoma development not only free melanoma cells from control by keratinocytes, but also provide new adhesion properties, resulting in switched partnerships with fibroblasts and vascular endothelial cells. The cadherin subtype switching also dictates gap junctional specificity in melanocytic cells during tumor development. This selective intercellular communication may contribute to the regulation of cell growth, differentiation, apoptosis, and migration of melanocytic cells in both physiologic and pathologic conditions. Abnormal up-regulation of the immunoglobin repeat-containing cell adhesion molecules Mel-CAM and L1-CAM potentiates invasion and migration of melanoma. Thus, abnormal expression of intercellular adhesion receptors and dysregulated intercellular communication underlies melanoma development and progression.
(1) Introduction
Generally, intercellular adhesion junctions utilize four structural types of adhesion receptors. The first group consists of cadherins: classic cadherins, which mediate cell-cell adhesion in the adherens junctions (Gumbiner, 2000); and desmosomal cadherins, which are present in desmosomes (Kowalczyk et al., 1999). The second structural type includes proteins with four transmembrane regions and intracellular amino and carboxyl termini. This diverse group consists of the gap junction proteins, connexins (Goodenough et al., 1996), and the tight junction proteins, occludins (Stevenson and Keon, 1998) and claudins (Morita et al., 1999). The third group includes those adhesion molecules with immunoglobulin-like repeats, such as Mel-CAM (MUC18, CD146) (Lehmann et al., 1989). The fourth group contains integrins. Although integrins are primarily involved in cell-matrix interactions (Hynes, 1992), they also participate in cell-cell adhesion. For example, integrin VLA-4 (α4 β 1) expression is characteristic of advanced primary tumors and mediates interaction of the tumor cells with vascular cell adhesion molecule-1 (VCAM-1) on vascular endothelium (Johnson, 1999).
Cutaneous melanoma is one of the fastest-rising malignancies in the last several decades (Ries et al., 2000). In contrast to many other cancer types, melanoma affects a relatively younger population and is notorious for its propensity to metastasize and for its poor response to current therapeutic regimens. Five steps of melanoma progression have been proposed based on clinical and histopathological features (Fig. 1) (Meier et al., 1998): (1) common acquired and congenital nevi with structurally normal melanocytes which have a finite lifespan and generally carry no cytogenetic abnormalities; (2) dysplastic nevi with structural and architectural atypia; (3) biologically early radial growth phase (RGP) primary melanoma, in which the cells have not yet metastasized; (4) advanced vertical-growth-phase primary melanoma (VGP), in which the cells have invaded the dermis and have the potential to metastasize; and (5) metastatic melanoma. The multi-stage nature of melanoma development makes it possible for us to follow the events for several intermediate stages and makes it an informative system for the investigation of changes in intercellular communication during oncogenesis.
This review will mainly focus on the kinetic properties of intercellular communication during melanoma development. Cadherins are major cell–cell adhesion molecules involved in the development and maintenance of skin (Takeichi, 1991; Gumbiner, 1996). Regulation of cadherin-mediated cell adhesion is thought to underlie the dynamics of the adhesive interactions between cells. Therefore, we will first discuss changes in cadherin-mediated adherens junctions in melanoma development. Then we will discuss the possible roles of other molecules such as connexins and immunoglobulin superfamily members in melanoma, although much less is known about those molecules.
(2) Cadherins in Normal Epidermal Development and Maintenance
There are three major cell types in human epidermis: keratinocytes, melanocytes, and Langerhans cells. The melanocyte in human skin is normally embedded in the basal layer of keratinocytes, anchored to the basement membrane of the epidermis. Each melanocyte extends its dendrites into the upper layers of the epidermis and establishes contacts with keratinocytes, forming the epidermal melanin unit that is comprised of 20 to 35 cells. In normal human skin, E-cadherin is expressed on the surfaces of all epidermal cells, including keratinocytes, melanocytes, and Langerhans cells, while P-cadherin is expressed only on the surfaces of basal layer keratinocytes (Tang et al., 1994). During human skin development, P-cadherin expression is spatiotemporally controlled and is closely related to the segregation of the basal layers as well as to the arrangement of epidermal cells into eccrine ducts (Furukawa et al., 1997; Jensen et al., 1997). N-cadherin is expressed in the human skin by dermal fibroblasts and vascular endothelial cells, but not by keratinocytes or melanocytes (Hsu et al., 1996).
During embryonic development, expression of the cadherin subtypes correlates with the migration and segregation of different cell layers and cell populations (Takeichi, 1995). Melanocytes and their progenitor melanoblasts are derived from the neural crest and migrate along the dorsolateral pathway to their final destination. In the mouse, at 11.5 days post-coitum, melanoblasts are in the dermis and are E-cad-P-cad-. During the next 48 hrs, a 200-fold increase of E-cadherin expression is induced on the surfaces of melanoblasts prior to their entry into the epidermis, thereby forming a homogeneous E-cadhighP-cad-/low population. The cadherin expression pattern then diversifies, giving rise to three populations, an E-cad-P-cad- dermal population, an E-cadhighP-cadlow epidermal population, and an E-cad-P-cadmed-high follicular population. In all three populations, the patterns of expression are region-specific, being identical to those of surrounding cells such as keratinocytes and fibroblasts. These findings suggest a role for E- and P-cadherins in guiding melanocyte progenitors to their final destinations (Nishimura et al., 1999), particularly during and after melanocyte entry into the epithelial layer, where the epidermal architecture of keratinocytes is maintained by E- and P-cadherins (Takeichi, 1988; Hirai et al., 1989a,b).
(3) E-cadherin is Critical for the Maintenance of Homeostasis in Epidermis
During melanoma development, a progressive loss of E-cadherin expression has been observed: Superficial compartments of nevi show heterogeneous membranous E-cadherin immunoreactivity (Silye et al., 1998), while junctional nevus cell nests display heterogeneous or diffuse cytoplasmic staining (Silye et al., 1998); melanoma cells, with few exceptions, do not express E-cadherin (Hsu et al., 1996; Scott and Cassidy, 1998). Despite the loss of E-cadherin expression by melanoma cells, these cells express high levels of N-cadherin both in vitro and in vivo (Li and Herlyn, 2000). The gain of expression of N-cadherin appears to be correlated with the progressive loss of E-cadherin. Nevus cells are generally heterogeneous for E-cadherin and N-cadherin, but more extensive studies need to be performed.
Down-regulation of E-cadherin expression or function is a critical factor in the malignant progression of most epithelial tumors. Disruption of E-cadherin-mediated cell adhesion facilitates tumor invasion, while re-establishing the functional cadherin complex—for example, by forced expression of E-cadherin—results in a reversion from an invasive to a benign phenotype (Guilford, 1999).
In normal human epidermis, E-cadherin is localized at the intercellular borders between keratinocytes and between keratinocytes and melanocytes (Tang et al., 1994; Karayiannakis et al., 1998). Loss of E-cadherin appears to be one of the critical steps in progression of melanoma, because loss of functional E-cadherin could trigger the release of cancer cells from the primary focus (Hirohashi, 1998). This process is probably due not to the loss of physical adhesion, but rather to multiple events that lead to uncontrolled proliferation and progressive invasion (Guilford, 1999).
The essential role of keratinocytes in the regulation of melanocyte growth and differentiation has been demonstrated (Donatien et al., 1993; Seiberg et al., 2000), but the molecule(s) that are responsible for the regulation are not yet well-understood. The studies by our group demonstrated that E-cadherin is the critical molecule in the control of melanocytes by keratinocytes (Hsu et al., 2000b). Normal melanocytes in monoculture in vitro have a phenotype similar to that of melanoma cells (Valyi-Nagy et al., 1993; Shih et al., 1994a). When the melanocytes are co-cultured with keratinocytes, expression of these melanoma-associated antigens, such as Mel-CAM and integrin αv β 3, is lost within 3-4 days, suggesting that the keratinocytes control the expression of cell-surface molecules on the melanocytes. Keratinocytes also control melanocyte proliferation. When keratinocytes and melanocytes are seeded together at a fixed ratio and allowed to proliferate, the original ratio remains constant during proliferation of both cell types, suggesting that the keratinocytes regulate their equilibrium with the melanocytes. The regulatory activity of keratinocytes occurs through direct cell-cell contact and not through soluble factors (Valyi-Nagy et al., 1993; Shih et al., 1994a). On the other hand, melanoma cells are refractory to regulation by keratinocytes (Valyi-Nagy et al., 1993; Shih et al., 1994a; Hsu et al., 2000b). Because melanoma cells generally do not express E-cadherin (Hsu et al., 1996; Scott and Cassidy, 1998), and the loss of regulatory dominance by keratinocytes occurs in concert with down-regulation of E-cadherin expression in melanoma cells, it was interesting to see whether forced E-cadherin expression in melanoma cells could restore keratinocyte-mediated growth control and down-regulate expression of invasion-related molecules. Our results show that E-cadherin expression in E-cadherin-negative melanoma cells leads to their adhesion to keratinocytes and renders them susceptible to keratinocyte-mediated control (Hsu et al., 2000b). After co-culture with keratinocytes, E-cadherin-expressing melanoma cells no longer express αv β 3 or Mel-CAM. In a skin reconstruction model, ectopic E-cadherin expression inhibits the invasion of melanoma cells into dermis (Hsu et al., 2000b).
(4) Regulation of Adherens Junctions
The regulation, under normal or malignant conditions, of the state of adherens junctions can occur at different levels and through different mechanisms (Gumbiner, 2000). Mutations in the E-cadherin gene and loss or mutation of the second, intact copy as well as mutation in the catenin genes, which encode proteins that interact with the cytoplasmic portion of E-cadherin, can be observed. In addition, transient or unregulated phosphorylation by receptor tyrosine kinases or non-receptor tyrosine kinases can interfere with the functional integrity of the adherens junction (Birchmeier et al., 1996).
A major form of regulation occurs by changing the level of cadherin gene expression, which influences the strength of adhesion (Steinberg and Takeichi, 1994). The type of cadherin expressed determines the specificities of cell interactions (Nose et al., 1988) and the properties of the interactions. Promoters of E- (Behrens et al., 1991; Ringwald et al., 1991; Bussemakers et al., 1994), P- (Faraldo and Cano, 1993; Faraldo et al., 1997), and N-cadherin (Li et al., 1997) have been isolated from different organisms. A palindrome element (E-pal) was shown to play an important role in the determination of the epithelial specificity of E-cadherin expression (Behrens et al., 1991; Hennig et al., 1996). E-pal is made up of two subsequent elements (E boxes CANNTG). E boxes are involved in the silencing of E-cadherin promoter activity occurring in cancer cells (Giroldi et al., 1997). However, no sequence homologous to E-pal has been found in the 5' region of P-cadherin (Faraldo and Cano, 1993) or N-cadherin (Li et al., 1997) gene. This could explain the differentially regulated expression of different cadherin subtypes during development and transformation. Another epigenetic mechanism that regulates E-cadherin expression is hypermethylation of cytosine–guanine (CpG) sites in regulatory regions (e.g., the promoter) (Graff et al., 1995; Yoshiura et al., 1995).
The strength and function of cadherin-mediated cell adhesion can be modulated rapidly in response to growth factors or other signals without gross changes in the expression level of the components of junctional complexes. This regulation is mainly accomplished by protein-protein interactions and post-translational modification of adhesive molecules. One important signaling pathway that regulates E-cadherin function involves the Rho family of small GTPases (RhoA, Cdc42, and Rac1), which regulate cell shape, growth, and polarity (Fukata et al., 1999; Kaibuchi et al., 1999). Rac1 and Cdc42, and their exchange factor TIAM 1 and target molecule IQGAP1 (Kuroda et al., 1998), regulate cadherin-mediated cell adhesion (Braga et al., 1997). IQGAP 1 appears to stimulate the dissociation of α-catenin from the E-cadherin cell adhesion complex by competing with α-catenin binding to β-catenin, which results in the loss or weakness of E-cadherin-mediated cell adhesion (Kuroda et al., 1998).
Tyrosine phosphorylation has also been implicated in the regulation of cadherin function. β-catenin can be tyrosine-phosphorylated by the non-receptor tyrosine kinase SRC, and this modification might lead to disassembly of the cadherin–catenin complex and the subsequent loss of cell adhesion (Behrens et al., 1993; Hamaguchi et al., 1993). Cadherin-mediated cell adhesion is regulated by tyrosine phosphatases in human keratinocytes (Soler et al., 1998). Moreover, both EGFR (epidermal growth factor receptor) and c-Met (scatter factor/HGF receptor) (DeLuca et al., 1999) phosphorylate β-catenin on tyrosine residues (Hoschuetzky et al., 1994), leading to inactivation of the E-cadherin/catenin complex through its interaction with β- or γ-catenin (plakoglobin) in the cytoskeleton (Jawhari et al., 1999). Hepatocyte growth factor/scatter factor (HGF/SF) has been shown to promote the de-epithelialization and migration of several cell types in vitro and in vivo (Stoker et al., 1987; Sonnenberg et al., 1993). c-Met is present in epithelial cells and melanocytic cells (Bottaro et al., 1991; Sonnenberg et al., 1993). In chick embryo epiblast cells, HGF/SF decreases the expression of E-cadherin and increases the percentage of cells with N-cadherin (DeLuca et al., 1999). Keratinocyte expression of transgenic HGF/SF affects melanocyte development, leading to dermal melanocytosis (Kunisada et al., 2000). Loss of E-cadherin expression in dermal melanocyte precursors suggests that HGF causes dermal localization of melanocytes and their precursors by down-regulation of E-cadherin molecules.
E-cadherin-mediated cell adhesion might also be abrogated through degradation of E-cadherin's extracellular portion by proteases such as stromelysin 1 (Lochter et al., 1997), which is activated during tumor progression.
Over-expression of ILK leads to a loss of cell-cell adhesion, which appears to be due to a dramatic decrease in E-cadherin expression, accompanied by the translocation of β-catenin to the nucleus (Novak et al., 1998; Wu et al., 1998). The expression of E-cadherin can be negatively regulated by LEF-1/β-catenin (Huber et al., 1996), thus providing a potential mechanism for the loss of adhesion.
Lately, it was found that the Snail family of transcription factors repress E-cadherin expression (Cano et al., 2000). Snail was originally implicated in the epithelial-mesenchymal transition required for the emigration of the neural crest from the neural tube and of the early mesoderm from the primitive streak during embryonic development (Nieto et al., 1994). Mouse Snail is a strong repressor of transcription of the E-cadherin gene, which specifically interacts with the E-pal element of the mouse E-cadherin promoter through its E2-box sequence (Batlle et al., 2000; Cano et al., 2000). Epithelial cells that ectopically express Snail adopt a fibroblastoid phenotype and acquire tumorigenic and invasive properties. Endogenous Snail protein is present in invasive mouse and human carcinoma cell lines and tumors in which E-cadherin expression has been lost.
(5) Cadherin Subtypes Determine Gap Junction Partnership
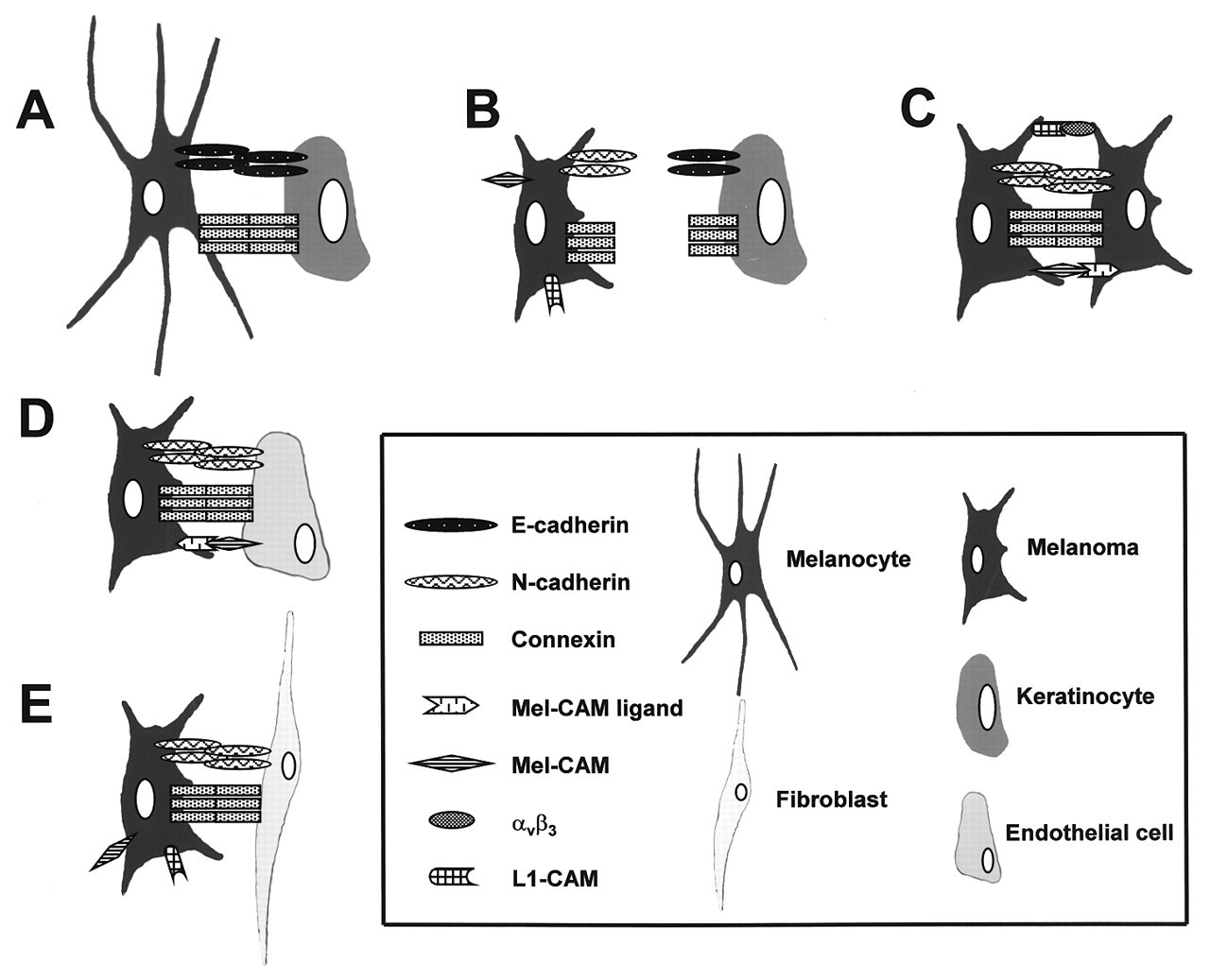
There are at least four groups of adhesion molecules that are involved in intercellular interactions (see above). These different types of interactions can cross-talk. To investigate the potential involvement of gap junctional perturbation in the pathogenesis of human melanoma, we studied intercellular communication in mono- and co-cultures of melanocytic and stromal cells using dye transfer assays (Hsu et al., 2000a). We found that melanocytes and melanoma cells have vastly different communication partner preferences. While melanocytes are compatible with their natural neighbors, the keratinocytes, melanoma cells exhibit communication capabilities among themselves and with dermal fibroblasts. The selective formation of heterotypic gap junctions between skin cells appears to be linked to cell-sorting by specific cadherins. Loss of E-cadherin and gain of N-cadherin expression during melanoma progression may endow melanoma cells with new communication and adhesion properties, facilitating gap junction formation with N-cadherin-expressing neighboring melanoma cells or dermal fibroblasts (Fig. 2). Restoration of E-cadherin expression in melanoma cells by adenoviral gene transfer resulted in re-establishment of gap junctional communication with keratinocytes. Analysis of our data suggested that cadherin-mediated cell-sorting and locus-specific membrane-docking dictate gap junctional specificity in melanocytic cells during tumor development. This selective intercellular communication may contribute to the regulation of growth and survival of melanocytic cells in both physiologic and pathologic conditions (Hsu et al., 2000a).
Gap junctions are clusters of integral membrane proteins, connexins (Cx). Six Cx subunits are radially arranged around a central pore, thereby forming a connexon assembly. The half-channels align and interact with complementary connexons present in the plasma membrane of neighboring cells (Krutovskikh and Yamasaki, 1997; Trosko and Ruch, 1998). This provides a direct pathway for small molecules (< 2 kDa), which may include ions (Ca2+, H+), secondary messengers (phosphatidyl inositides, cAMP), and metabolic products (amino acids), between the coupled cells. Specificity of gap junction allows for the formation of communication compartments essential for tissue function and homeostasis. Reduced or lost gap junctional activity has been implicated in various human cancers, such as squamous cell carcinoma (Tada and Hashimoto, 1997), lung cancer (Jinn et al., 1998), gastric carcinoma (Uchida et al., 1995), hepatocellular carcinoma (Krutovskikh et al., 1994), glioblastoma (Huang et al., 1999), and prostate cancer (Tsai et al., 1996). Exogenous expression of Cxs in Cx- and gap junction-deficient cell lines restores functional communication, which in turn retards tumor growth both in vitro and in vivo (Eghbali et al., 1991; Hirschi et al., 1996; Huang et al., 1998). While these results suggest that connexin genes form a family of tumor suppressor genes, some neoplastic cells have normal or even greater gap junction expression and cell-cell coupling (Trosko and Ruch, 1998) or, in melanoma, altered partners (Hsu et al., 2000a). They may have functional gap junctions among themselves, or with stromal cells, but are incapable of gap junction formation with non-transformed cells. Therefore, the tumor cells might be isolated from the regulatory influence from their surrounding normal cells. This deficiency in communication might be due to differences on the cell surface—for example, cell-cell adhesion molecules such as cadherins—that fail to provide the appropriate cell-cell contact needed for gap junction (Kanno et al., 1984; Mege et al., 1988; Meyer et al., 1992; Frenzel and Johnson, 1996; Fujimoto et al., 1997). The altered expression profile of cadherin molecules and the subsequent switch to atypical gap junction partners leading to abnormal gap junction communication between malignant cells and host cells could underlie the dysregulated proliferation and invasion of tumor cells. For example, when B16 mouse melanoma cells were transfected with wild-type Cx26, the resulting cells became competent for coupling with endothelial cells and more competent for metastasis, suggesting that certain connexin(s) play a role in intravasation and extravasation of tumor cells through heterologous gap junction formation with endothelial cells (Ito et al., 2000).
At present, the mechanisms as to how the different heterotypic gap junctions serve to coordinate epidermal morphogenesis and melanocytic transformation remain unclear. There is emerging information showing that gap junctions serve to regulate cell growth and tissue morphogenesis. For example, gap junctional intercellular communication (GJIC) can modulate gene expression, presumably via the modulation of signals that diffuse from cell to cell (Huang et al., 1998). It is conceivable that alterations in gap junction signaling may directly stimulate melanomagenesis or simply allow for tumor progression toward a more malignant phenotype through the loss of normal homeostatic growth regulation. Further elucidation of the molecular components of the partner-specific gap junctional signaling pathways in skin cells may provide new insights into the pathogenesis of human melanoma.
There are extensive cross-talks between and among different forms of cell adhesion. Cadherin-mediated cell adhesion appears to be the most critical one, because it can, at least in some cases, initiate the formation of other cell-cell junctional complexes, including tight junctions (Siliciano and Goodenough, 1988), desmosomes (Pasdar and Nelson, 1988a,b), and gap junctions (Kanno et al., 1984; Mege et al., 1988; Meyer et al., 1992; Frenzel and Johnson, 1996; Fujimoto et al., 1997) (see above). However, it is not clear whether desmosomes or tight junctions are present in melanocyte-keratinocyte units and whether they play a role in melanoma development and progression.
(6) CAMs in Melanoma Development
During melanoma development, the expression of several immunoglobin repeat-containing cell adhesion molecules (CAMs) is also a dynamic process. Two molecules, Mel-CAM and L1-CAM, are among the most investigated.
The cell-cell adhesion protein Mel-CAM (MUC18, CD146), a member of the immunoglobulin superfamily (Lehmann et al., 1989; Sers et al., 1993), has been strongly implicated in melanoma progression and metastasis (Lehmann et al., 1987). Mel-CAM mediates melanoma cell interactions via heterotypic Ca2+-independent adhesion to a currently undefined ligand (Shih et al., 1997a,b). Although not expressed on normal melanocytes in vivo, and only rarely detected on benign nevus cells, Mel-CAM is highly expressed in most metastatic melanoma lesions and advanced primary tumors (Shih et al., 1994b). In addition, Mel-CAM expression is up-regulated proportional to increasing vertical tumor thickness, which is an established indicator of metastatic potential (Johnson, 1992). Mel-CAM-negative melanoma cells with a non-metastatic and low tumorigenic profile become highly tumorigenic and harbor increased metastatic potential upon transfection with Mel-CAM in vivo (Luca et al., 1993; Xie et al., 1997). Since Mel-CAM expression correlates closely with increased tumor growth and metastasis, this protein is very likely to play an important role in defining the melanoma phenotype (Johnson et al., 1997; Shih et al., 1997a). The observation that the expression of both Mel-CAM and its ligand seems to be required for tumor progression in melanoma suggests that this adhesion system can provide melanocytic cells with the necessary cell-cell interaction properties to enhance tumorigenicity.
The cell adhesion molecule L1 (L1-CAM) is a transmembrane glycoprotein with six immunoglobulin-like (Ig) and five fibronectin-type-III-like (FN III) domains in its extracellular segment. L1-CAM functions not only as an adhesive molecule but also as a signal-transducing receptor (Schuch et al., 1989). L1-CAM cytoplasmic domain plays a significant role in signal transduction and interactions with the cytoskeleton (Davis and Bennett, 1994; Wong et al., 1996; Dahlin-Huppe et al., 1997). Thus, L1-CAM can influence cell growth and migration behavior in response to ligand binding. L1-CAM mediates neuronal adhesion and fasciculation, and stimulation of fibroblast growth factor (FGF)-receptor-dependent neurite outgrowth by homophilic interaction. Recent findings also revealed heterophilic interactions between L1-CAM and matrix receptors such as human integrin αV β 3. The integrin-binding site of L1-CAM contains the tri-peptide Arg-Gly-Asp (RGD). L1-CAM-integrin binding predominates in leukocyte subsets and in several tumors. It can mediate homotypic and heterotypic cell-cell adhesion and cell movement on substrate-embedded L1-CAM. Its functional versatility can be attributed to its diverse choice of ligands, presumptive modulation by co-expressed surface molecules, and alternative signal transduction pathways.
A potential function for L1-CAM in tumor progression is suggested by its widespread expression on many tumor cells, including melanoma (Mujoo et al., 1986; Wolff et al., 1988; Linnemann et al., 1989; Kobayashi et al., 1991), lung carcinomas (Katayama et al., 1997), and monocytic leukemias (Pancook et al., 1997). Elevated levels of L1-CAM on a metastatic variant of a melanoma cell line suggested a role for L1-CAM in tumor progression (Linnemann, et al., 1989). L1-CAM may promote metastasis by facilitating tumor cell invasion or migration (Ohnishi et al., 1998). However, the exact role of this molecule in invasion and metastasis in melanocytic transformation still needs to be determined.
From a differential display comparing mRNA populations isolated from a non-metastatic and highly metastatic human melanoma cell line, the activated leukocyte cell adhesion molecule (ALCAM) was identified (Degen et al., 1998). ALCAM is involved in homophylic (ALCAM-ALCAM) and heterophylic (ALCAM-CD6) cell adhesion interactions. Expression of ALCAM correlates with the aggregation and metastatic capacity of human melanoma cell lines in vitro (Degen et al., 1998). Immunohistochemistry on human melanocytic lesions also revealed that ALCAM expression correlates with melanoma progression in vivo. Most nevi and RGP melanomas studied did not express ALCAM, while some VGP melanomas did. The fraction of positive lesions further increased in higher progression stages (van Kempen et al., 2000).
(7) Future Research
We are beginning to understand the dynamics of tissue and organ morphogenesis through cell-cell and cell-matrix communications. The human skin has a unique architecture that is obviously different from that of the mouse. Thus, a knock-out or transgenic model cannot provide faithful information. Therefore, the better models would be human skin reconstructions (in vitro) and human skin xenografts on the mouse (in vivo). These models will allow us to investigate normal skin morphogenesis and alterations in melanoma development and progression.
It remains to be studied how tumor cells reconcile their requirements for variations in cell adhesion, i.e., down-regulation of E-cadherin activity, to break away from the primary tumor site followed by involvement of other adhesion molecules in cell-substrate and cell-cell interactions during metastasis. The diverse requirements might be met by switching classes or subtypes of adhesion receptors that favor different biological processes.
E-cadherin-mediated adhesion between melanocytes and keratinocytes is critical for intercellular signaling. Even highly aggressive metastatic melanoma cells can be signaled to shut off expression of genes associated with tumor invasion and metastasis, suggesting that this strategy could be utilized for melanoma therapy. However, it remains to be determined whether the signals between the cells are transmitted through E-cadherin only or through another cell-cell adhesion system, i.e., a co-receptor. It has been controversial whether the loss of E-cadherin-mediated cell adhesion is a prerequisite for tumor progression, or whether it is instead a consequence of de-differentiation during tumor progression in vivo. The loss of cell–cell adhesion alone is not sufficient to induce active tumor invasion and metastasis; additional, genetic or epigenetic, events seem to be involved. It is likely that N-cadherin promotes a state of dynamic adhesion that allows for both attachment and detachment of individual cells from the primary tumor and selective association with critical tissues such as the stroma and the endothelium.
The biological events of melanocyte proliferation in normal skin are unknown. Melanocytes embedded in the epidermis among the basal layer keratinocytes are not proliferative but maintain the capacity to do so. When the total surface area of skin increases, melanocytes may proliferate at a rate which keeps the ratio of melanocytes to keratinocytes constant. How this process is regulated is still a mystery.
Overall, the ultimate aim of the studies is to understand the mechanisms of melanoma initiation and progression, and to find ways to prevent and cure the disease. Because cancers are regarded as diseases caused by the disruption of homeostasis, re-establishing homeostasis is a logical approach to reversing the malignancy.
Dynamic expression of cell adhesion molecules during melanoma development. Dynamics of cell interactions and communication during melanoma development. 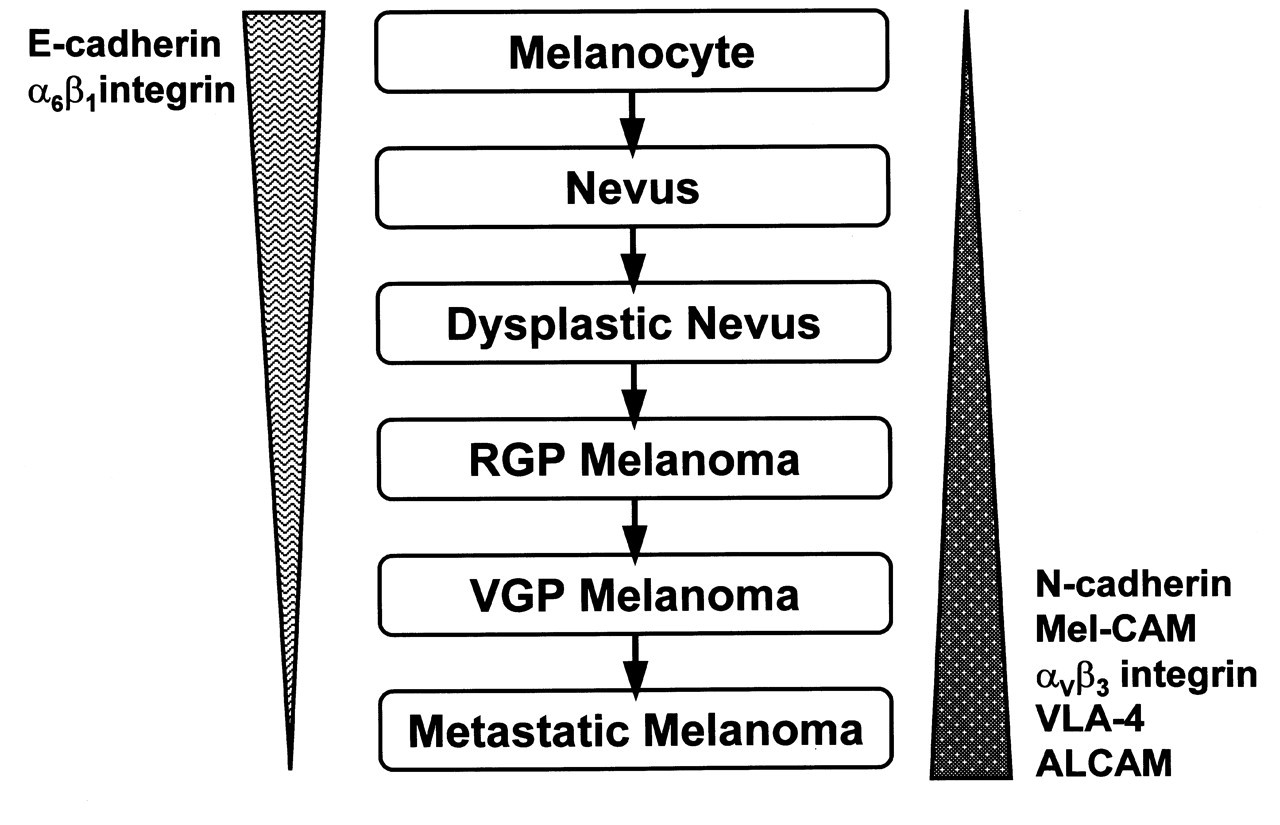
Footnotes
Acknowledgements
We thank all members in Dr. Meenhard Herlyn's lab for their invaluable support and advice. This work was supported by NIH grants CA-25874, CA-76674, CA-47159, CA-80999, and CA-10815.