
Abstract
The dysregulation of the molecular events governing cell cycle control is emerging as a central theme of oral carcinogenesis. Regulatory pathways responding to extracellular signaling or intracellular stress and DNA damage converge on the cell cycle apparatus. Abrogation of mitogenic and anti-mitogenic response regulatory proteins, such as the retinoblastoma tumor suppressor protein (pRB), cyclin D1, cyclin-dependent kinase (CDK) 6, and CDK inhibitors (p21WAF1/CIP1, p27KIP1, and p16INK4a), occur frequently in human oral cancers. Cellular responses to metabolic stress or genomic damage through p53 and related pathways that block cell cycle progression are also altered during oral carcinogenesis. In addition, new pathways and cell cycle regulatory proteins, such as p12DOC-1, are being discovered. The multistep process of oral carcinogenesis likely involves functional alteration of cell cycle regulatory members combined with escape from cellular senescence and apoptotic signaling pathways. Detailing the molecular alterations and understanding the functional consequences of the dysregulation of the cell cycle apparatus in the malignant oral keratinocyte will uncover novel diagnostic and therapeutic approaches.
(I) Introduction
Dysregulation of the cell cycle machinery is a fundamental hallmark of cancer progression (Lundberg and Weinberg, 1999). The cellular programs of proliferation, differentiation, senescence, and apoptosis are intimately linked to the cell cycle regulatory machinery. Many of the molecular alterations that cause abnormal biologic behavior of cancer cells are based on aberrations of cell cycle regulation. For example, escape from dependence on mitogens or induction of resistance to anti-mitogens, tolerance to DNA damage, apoptosis resistance, and progression of cells with activated oncogenes and/or inactivated tumor suppressor genes through multiple checkpoints resulting in increased genomic instability—all affect and/or are affected by cell cycle regulatory proteins (Lundberg and Weinberg, 1999).
We are beginning to understand the dysregulation of cell cycle progression during oral carcinogenesis. The signaling pathways stimulated by exogenous and endogenous signals will be discussed as they relate specifically to oral keratinocyte biology. In addition, we will explore the potential clinical impact of this knowledge and highlight some important questions that remain regarding the contribution of cell cycle dysregulation during oral carcinogenesis.
(II) Extracellular Signaling and Cell Cycle Regulation
(A) The cell cycle clock
The cyclins and cyclin-dependent kinases (CDKs) form the core of cell cycle regulation (Fig. 1) (Sherr, 2000). Expression of cyclins is cell cycle phase-dependent and is regulated transcriptionally, post-transcriptionally, and translationally/post-translationally. The cyclin family members interact with CDKs, and these complexes are required to pass through specific phases of the cell cycle. D-type cyclins interact with CDK4 and CDK6 and are necessary for G0/G1 transition. Cyclin E binds to CDK2 and mediates S phase entry. The cyclin A/CDK2 complex regulates passage through the S-phase. Later, in conjunction with CDC 2, cyclin A also induces the G2 phase of the cell cycle. Cyclin B1 and CDC2 trigger the molecular events associated with mitosis (Lundberg and Weinberg, 1999).
In contrast to the cyclins, the steady-state levels of CDKs are not markedly modulated during the cell division cycle. The cyclins complex with the CDKs and activate their catalytic activities, and these cyclin/CDK complexes each phosphorylate-specific sets of target proteins that regulate cell cycle progression.
(B) The retinoblastoma tumor suppressor and regulation of the restriction point
The decision to activate the cascade of events leading to DNA synthesis and ultimately cell division occurs at the restriction (R) point. The sum of signals accumulated in the first two-thirds of the G1 phase may trigger cell cycle progression, cause the cell to revert to G0 (or quiescence), or lead to cellular differentiation. After passing the R point late in G1, a cell will disregard exogenous signals and will enter DNA synthesis. Thus, the molecular mechanisms that control progression through the R point are of central importance in governing entry into the cell cycle. After DNA synthesis, major intracellular insults such as genomic damage or metabolic disruption can halt cell cycle progression, and the cells will arrest at other checkpoints in the S, G2, or M phase.
The retinoblastoma protein (pRB) is likely a key switch at the R-point (Lundberg and Weinberg, 1999) (Fig. 1). Phosphorylation results in structural conformation of proteins that initiate or inhibit cell physiologic events. Cells arrest near the R-point when pRB is hypophosphorylated. Upon pRB phosphorylation, cells advance to late G1 and subsequently undergo DNA synthesis. Initial pRB phosphorylation events are mediated by cyclin D1/CDK4 and/or cyclin D1/CDK6 complexes. Cyclin E/CDK2 additionally phosphorylates pRB during entry into and progression through the S-phase (Hinds et al., 1992, 1994; Sherr, 1993). Hypophosphorylated pRB inhibits S phase entry by physically associating with members of the E2F transcription factor family, a group of five members (E2F1-5). E2F1-3 preferentially bind to pRB; E2F4 and 5 bind to pRB-related proteins p107 and p130. The E2F-pRB complex actively represses transcription of target genes that regulate DNA synthesis (Nevins, 1998). Upon pRB phosphorylation, pRB/E2F complexes are disrupted, and E2F can now transcriptionally activate these promoters. Genes transcriptionally activated by E2F family members binding include c-MYC, n-MYC, CDC-2, p21WAF-1, cyclin A c-MYB, and the epidermal growth factor receptor (Goodger et al., 1997). Over-expression of E2F-1 has been shown to reverse the effects of telomerase activity in head and neck cancer cell lines (Henderson et al., 2000). Therefore, pRB or members of its regulatory pathway become critical molecular targets for malignant progression.
The function of pRB is lost in all retinoblastomas, a relatively rare form of hereditary childhood eye tumor. In addition, mutations in pRB due to somatic inactivation are also found in a variety of other tumors, including sarcomas, small-cell lung carcinomas, and bladder cancers. Re-introduction of the RB cDNA into such cancer cells inhibits their proliferation and tumorigenic properties, thus supporting the critical role of pRB-inactivating mutations in the formation of neoplastic cells (Weinberg, 1995). A central role for pRB in the genesis of human tumors is also suggested by the observation that pRB is targeted by the oncoproteins encoded by several DNA tumor viruses, including the human papilloma virus (HPV) E7 oncoprotein (Dyson and Harlow, 1992). Many other tumors, including those of the breast, prostate, and brain, however, show only infrequent loss of pRB expression (Weinberg, 1995). The delineation of the signal transduction pathways that govern pRB activity has suggested that pRB function may also be abrogated by mutations of specific components in this pathway. This concept has been validated, and it is now recognized that almost every human tumor has sustained an aberration in the pRB pathway. Interestingly, specific alterations in the pRB pathway are preferentially observed in individual tumor types, suggesting that there is additional complexity in this pathway.
Although pRB mutations are rare in oral cancer, the pRB regulatory circuit is also abrogated in these tumors. Several reports suggest that pRB function is absent in malignant oral epithelium (Pavelic et al., 1996; Yokoyama et al., 1996; Saito et al., 1999; Schoelch et al., 1999). Studies demonstrating reduced pRB expression frequently did so only in tumors with normal p16INK4a (El-Naggar et al., 1999; Lai and El-Naggar, 1999; Sartor et al., 1999). Other reports demonstrate elevated pRB levels during oral cancer progression, suggesting that pRB overexpression may be protective against apoptosis (Regezi et al., 1999). Altered expression was found to be associated with clinically aggressive oral cancers and tobacco/betal quid use, but did not correlate with early (< 35 yrs) or late (> 75 yrs) onset of disease (Pavelic et al., 1996; Pande et al., 1998; Regezi et al., 1999). While the direct contribution of altered pRB expression during oral carcinogenesis has not been completely defined, there is mounting evidence that the pRB pathway is rendered dysfunctional in oral cancers by abrogating upstream-positive (cyclin D1 and CDK6) or -negative (p21WAF1, p27KIP1, p16INK4a, and p19ARF) regulators.
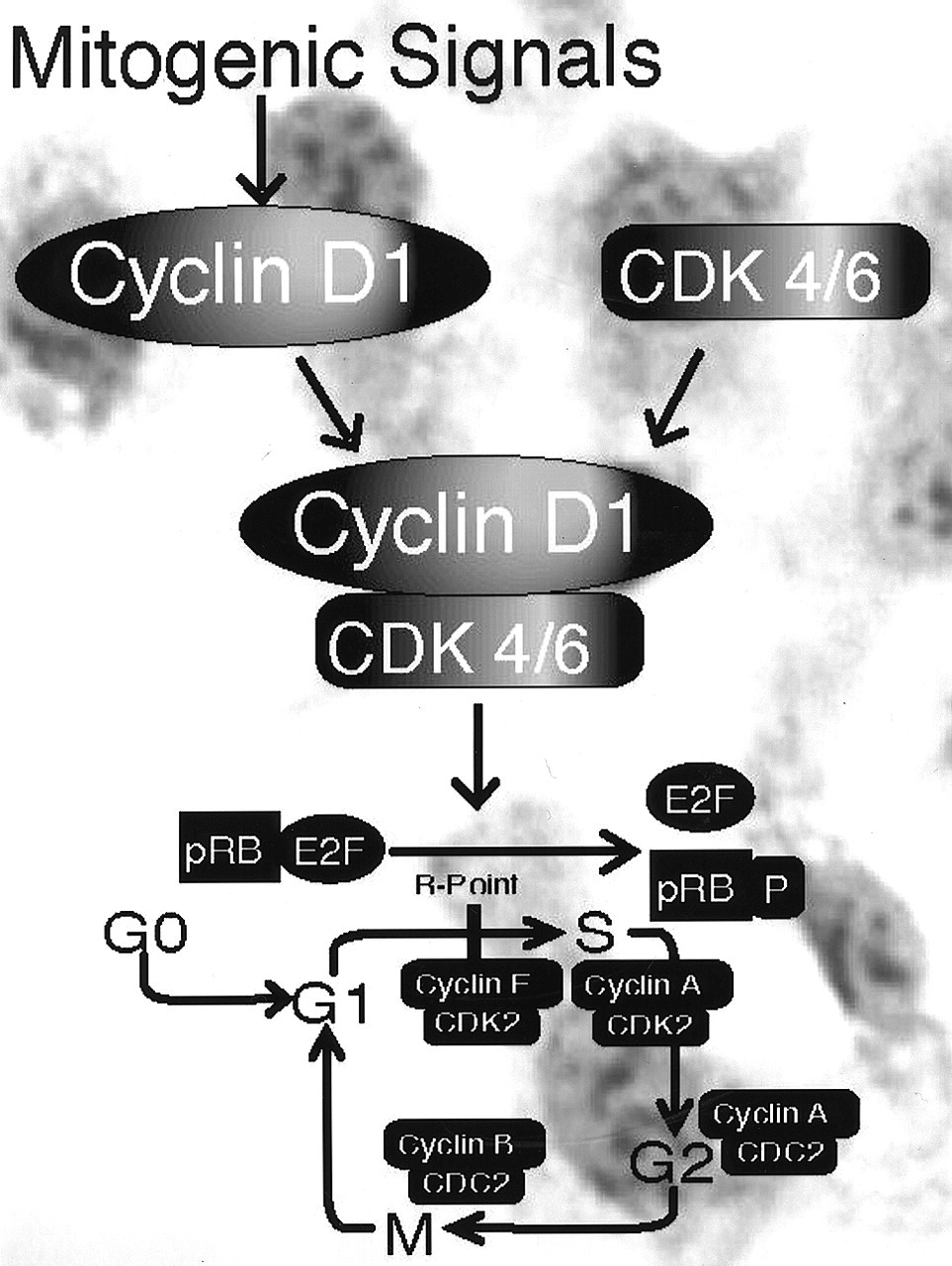
(C) Cyclin D1 and mitogen-activated cell cycle progression
D-type cyclins represent a link between upstream mitogenic stimuli and regulation of pRB function (Sherr, 1993, 1994a) (Fig. 2). Human cyclin D1 was first isolated in human parathyroid adenomas as a gene rearranged by translocation to the parathyroid hormone locus at 11q13 (Motokura et al., 1991). Two other human D-type cyclin genes, cyclins D2 and D3, have also been cloned. All three human D-type cyclin genes encode 33-34-kDa proteins that share an average of 57% identity over the entire coding region and 78% in the cyclin box, the region of the cyclins that interacts with CDKs. There is compelling evidence for a role of cyclin D1 in G1 phase progression in the cell cycle. Microinjection of cyclin D1 antibody or antisense cyclin D1 blocks cells from entering the S phase (Baldin et al., 1993). Overexpression of cyclin D1 accelerates progression through the G1 phase of the cell cycle and reduces the requirement of the cell for mitogens (Quelle et al., 1993). D-type cyclins and their catalytic partners, CDK4 and CDK6, play an important role in modulating the response to extracellular stimuli and are believed to be essential for passage through the G1 phase (Sherr, 1993).
As indicated earlier, pRB and pRB-related proteins (p107, p130) are important physiological substrates of CDK4/cyclin D and CDK6/cyclin D complexes, although other substrates likely exist (e.g., myb-like transcription factor, or mib) (Neuman et al., 1997; Stepanova et al., 2000). Phosphorylation of pRB by cyclin D/CDK4/6 complexes occurs late during the G1 phase and abrogates its growth-inhibitory function.
Cyclin D1's role as an oncogene has been established by its ability to cooperate with RAS or complement a defective adenoviral E1a oncogene in cell transformation assays (Hinds et al., 1994; Robles et al., 1996, 1998; Rodriguez-Puebla et al., 1999a,b). Overexpression of cyclin D1 has been reported in a variety of human tumors, including breast carcinomas, mantle cell lymphomas, and squamous cell carcinomas derived from the oral cavity, larynx, and esophagus as well as from other sites (Sicinski et al., 1995). The mechanisms underlying cyclin D1 overexpression in cancer include gene amplification, chromosomal translocation, and mitogenic stimulation of gene transcription (Hinds et al., 1994; Sherr, 1994a). The abrogation of mitogenic pathways that might lead to overexpression of cyclin D1 during oral carcinogenesis has been reviewed extensively elsewhere (Kamer et al., 1999). In the majority of primary oral cancers and cell lines, cyclin D1 overexpression appears independent of p16INK4a inactivation (Okami et al., 1999). The role of cyclins D2 and D3 in oral carcinogenesis is poorly understood. Expression of antisense cyclin D1 induces apoptosis and tumor shrinkage in squamous cell carcinomas (Sauter et al., 1999). Moreover, cyclin D1 overexpression has also been linked to increased risk of occult metastases and poor prognosis in oral cancer patients (Capaccio et al., 2000).
Animal models are currently being used to elucidate the contribution of cyclin D1 to human development and carcinogenesis (Bodrug et al., 1994; Lovec et al., 1994; Fantl et al., 1995; Mueller et al., 1997; Soonpa et al., 1997; Ma et al., 1998). Expression of the human cyclin D1 gene from the MMTV promoter caused mammary hyperplasia and carcinomas in lactating transgenic mice (Wang et al., 1994). When expression of human cyclin D1 is targeted to the oral-esophageal squamous epithelium with the EBV ED-L2 promoter, the ED-L2 cyclin D1 transgenic mice develop severe oral dysplasia. Expression of cyclin D1 from a keratin promoter (K5) also causes similar lesions (Nakagawa et al., 1997). The targeting of the cyclin D1 oncogene by an Epstein-Barr virus promoter in transgenic mice causes dysplasia in the tongue, esophagus, and forestomach (Mueller et al., 1997; Nakagawa et al., 1997). A transgenic mouse model with cyclin D1 overexpression results in cell cycle, epidermal growth factor receptor, and p53 abnormalities (Mueller et al., 1997). Ongoing work suggests that when the ED-L2 cyclin D1 transgenic mice are bred with mice in which the p53 gene product is ablated, the compound or hybrid mice show accelerated oral squamous dysplasia which progresses to oral squamous cancer (Opitz et al., unpublished observations). Thus, in this mouse model of oral carcinogenesis, cyclin D1 overexpression and p53 inactivation cooperate. Given the parallels to human oral carcinogenesis, this model will be useful for the testing of chemopreventive and therapeutic agents.
Altered CDK6 expression has also been detected in an oral squamous cell carcinoma (SCC) cell line (Timmermann et al., 1997). Further analysis of a set of human oral SCC lines showed that activity of CDK6 was increased in these lines in the absence of cyclin D1 overexpression (Timmermann et al., 1997). The specific hyperactivity of CDK6 found in these oral cancer cell lines may reflect a unique role for CDK6 in promoting proliferation in these cells that cannot be fulfilled by CDK4 (Timmermann et al., 1997). Indeed, a role of CDK6 in promoting G1 transit has also been observed in cultured osteosarcoma cells, and may involve both spatial regulation and target specificity of this kinase (Grossel et al., 1999). In any case, it is clear that loss of p16INK4a expression, usually by methylation of the promoter or by deletion, is a common mechanism for CDK6 activation and dysregulation of the pRB pathway in oral carcinoma cell lines (Timmermann et al., 1997, 1998).
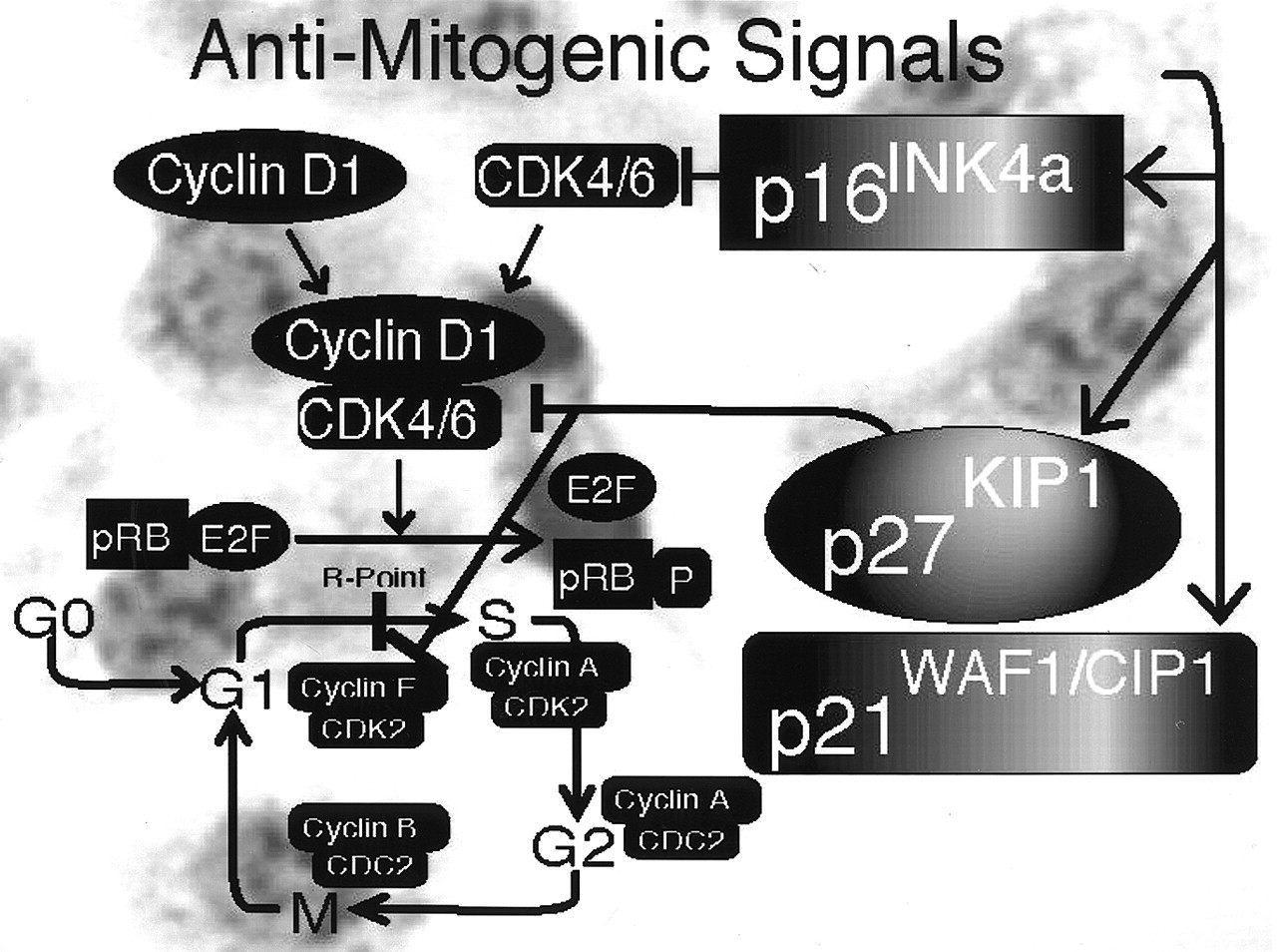
(D) Regulation of R-point by anti-mitogenic extracellular factors
Exogenous anti-mitogenic signals also converge on the pRB regulatory pathway (Fig. 3). Various extracellular signals, such as growth factor deprivation or the TGF-β signaling cascade, inhibit cellular proliferation by inhibiting CDK activity through CDK inhibitors (CDKi). Mitogenic stimuli also appear to act on CDKis. The CIP/KIP family of CDKis (p21WAF1/CIP1, p27KIP1, p57KIP2) can inhibit any cyclin/CDK complex. In addition, binding of p21WAF1/CIP1 to the CDK2-proliferating cell nuclear antigen results in slowing of DNA replication (by inhibiting the elongation of the strand being polymerized and allowing repairs to be made to damaged DNA) (Goodger et al., 1997). In addition, the CIP/KIP family members also play an important role in the assembly of functional CDK4 and CDK6/cyclin D complexes. In contrast, the INK family of CDKis (p15INK4b, p16INK4a, p18INK4c, p19INK4d) specifically inhibits CDK4 and CDK6 by disrupting active kinase complexes with cyclin D1, and does not inhibit other CDKs.
Down-regulation of CDKis has been described in oral cancers. Loss of p16INK4a function is frequently observed in a variety of human cancers, including oral and esophageal cancers (Timmermann et al., 1997, 1998; Pande et al., 1998; El-Naggar et al., 1999; Sartor et al., 1999; Schoelch et al., 1999). p16INK4a is one of the proteins encoded by the MTS-1 tumor suppressor locus on chromosome 9p. Inactivation of this locus can be the result of chromosomal deletion, point mutation, or promoter hypermethylation (Akanuma et al., 1999; Cody et al., 1999; Miracca et al., 1999; Sanchez-Cespedes et al., 2000). Reduced p16INK4a function was frequently found in pRB-positive oral tumors (Pande et al., 1998; Timmermann et al., 1998; El-Naggar et al., 1999; Sartor et al., 1999). p16INK4a inactivation is believed to be an early event in oral carcinogenesis that is related to loss of replicative senescence/ immortalization (Loughran et al., 1996; Papadimitrakopoulou et al., 1997; Rocco et al., 1998; Timmermann et al., 1998; Chen et al., 1999). Loss of p16INK4a function is correlated with a poor prognosis in oral cancer patients (Bova et al., 1999; Danahey et al., 1999). A reduction of p27KIP1 during oral carcinogenesis has also been reported (Schoelch et al., 1999; Kudo et al., 2000a). Overexpression of p27KIP1, either experimentally in oral cancer cell lines using a proteasome inhibitor or as observed by inmmunohistodetection in human oral cancer biopsies, appears to correlate with the induction of apoptosis (Fujieda et al., 1999; Kudo et al., 2000b). Reduced p27KIP1 levels have been correlated with unfavorable treatment response, shorter overall survival, and shorter disease-free survival (Mineta et al., 1999; Venkatesan et al., 1999). Forced expression of p21WAF1/CIP1 in human squamous cell carcinoma xenografts in athymic mice has been shown to retard cell proliferation (Cardinali et al., 1998). In oral tumors, proliferating malignant oral keratinocytes have been demonstrated to co-express cyclin D1, suggesting that the latter can override p21WAF1/CIP1 inhibition of cell cycle progression (van Oijen et al., 1998). Reduced expression of p21WAF1/CIP1 appears to correlate with poor patient survival better than does reduced p53 expression (Tatemoto et al., 1998; Kudo et al., 1999). Interestingly, no correlation has been found between p53 and p21WAF1/CIP1 expression, suggesting that p21WAF1/CIP1 expression may rely on p53-dependent and p53-independent pathways (Yook and Kim, 1998; Kudo et al., 1999).
(III) Cell Cycle Regulation Response to Intracellular Events
Cell cycle progression may be delayed or blocked by genotoxic and metabolic insults. Signaling pathways, such as p53, can govern both the arrest of cell division and the initiation of an apoptotic response, dependent on the cellular damage. Therefore, cellular pathways such as differentiation, senescence, and apoptosis converge with, and are frequently activated by, inhibitors of cell division.
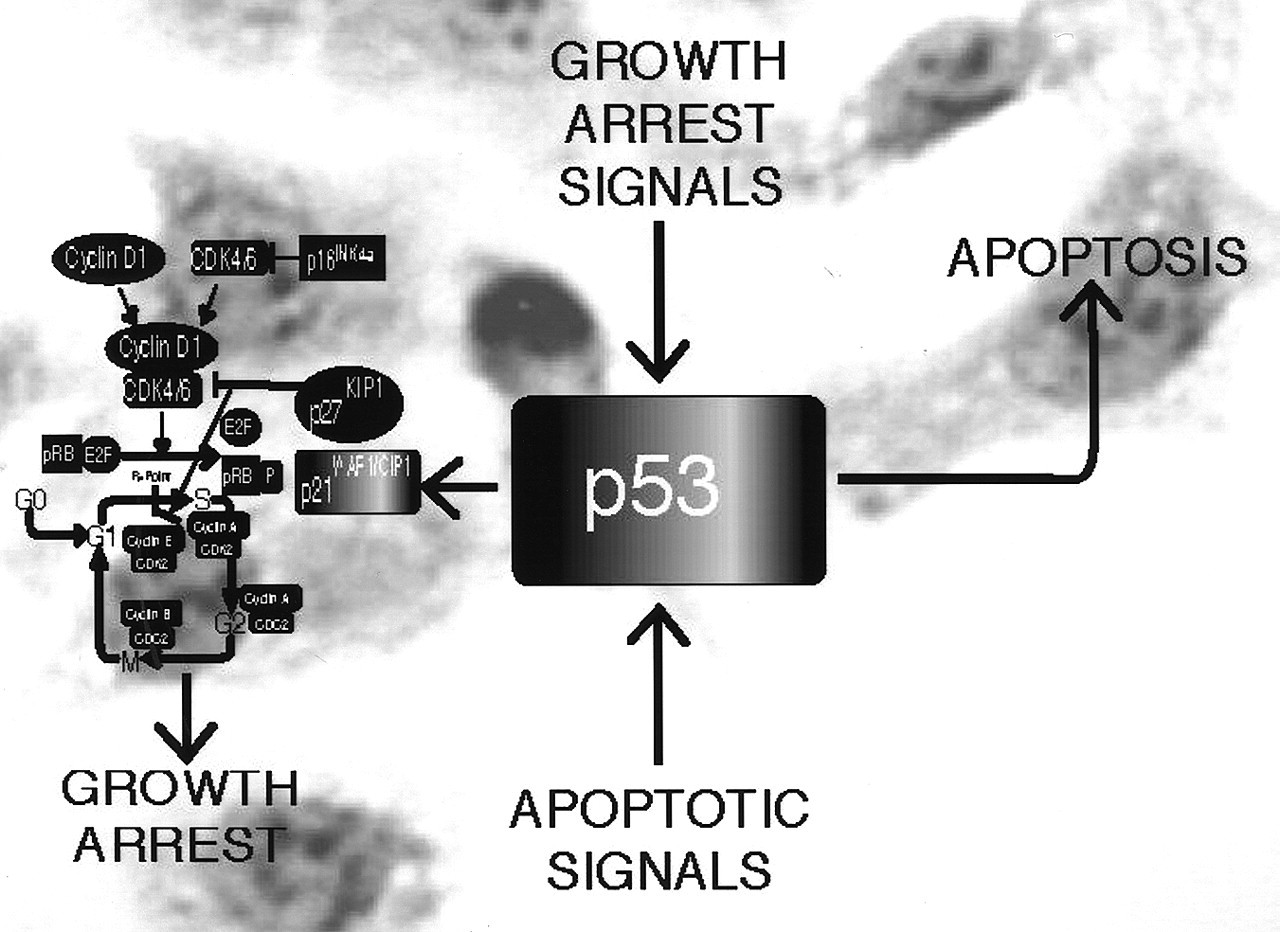
(A) The p 53 pathway and genomic damage
Genomic damage and other cellular stress signals elicit a cellular response pathway that delays or prevents cell division (Fig. 4). The p53 tumor suppressor protein acts as a master regulator of this pathway and induces cell cycle inhibition and/or apoptosis following DNA damage. Under normal cellular growth conditions, p53 has a short half-life, and the cellular steady-state levels are very low. When treated with a variety of DNA-damaging agents, such as ultraviolet or ionizing radiation and certain chemotherapeutic drugs, normal cells respond by a rapid, non-transcriptional induction of p53 as well as post-translational modification including phosphorylation through the ATM/ATR and DNA PK family of protein kinases (Giaccia and Kastan, 1998). Depending on the circumstances, this induction of p53 causes cell cycle arrest or apoptotic cell death (Prives, 1993). The p53-mediated induction of apoptosis is the subject of intense research and involves both transcriptional and non-transcriptional mechanisms (Caelles et al., 1994; Dynlacht et al., 1994, 1997; Qin et al., 1994; Krek et al., 1995; Miyashita and Reed, 1995).
The induction of G1 growth arrest by p53 is mediated by the transcriptional activity of p53. Expression of p21WAF1/CIP1 mRNA and protein is strongly induced by p53 under these conditions, and p21WAF1/CIP1 can itself inhibit cell proliferation upon introduction into cultured cells (El-Deiry et al., 1993; Harper et al., 1993). The ability of p21WAF1/CIP1 to inhibit many different cyclin/CDK complexes suggests that p21WAF1/CIP1 has a broad effect on cell cycle progression (Sherr, 1994b). This p53-mediated cell cycle arrest in response to DNA damage is thought to prevent the perpetuation of genomic mutations by allowing a cell to repair DNA damage before it undergoes a new round of DNA replication. Thus, p53 is involved in maintaining the integrity of the genome and has been referred to as the 'guardian of the human genome' (Lane, 1992). Moreover, it is widely believed that the common absence of functional p53 in human tumors contributes to genomic instability, which is a hallmark of human tumors and pathologically manifested as nuclear pleomorphism.
(B) The p 53 pathway and aberrant cell physiology
The p53 and pRB tumor suppressor pathways are linked in many ways. On one hand, the ability of p53 to induce G1 growth arrest critically depends on the integrity of pRB (Lundberg and Weinberg, 1999). Conversely, abnormalities in the pRB pathway are sensed by p53. Normally, E2F activity is tightly regulated by pRB. Dysregulated E2F activity, for example, caused by a mutation in the pRB pathway triggers apoptosis that is at least in part p53-mediated.
Loss of p53 function is well-documented in oral cancers, and p53 mutations have been reported in over 60% of oral squamous cell carcinomas (Sakai et al., 1992). Mutation of p53 frequently induces a stabilization of the mutated protein. While little p53 is detected in normal oral epithelia and low-grade leukoplakias (mild to > moderate dysplasia), p53 accumulation (indicative of p53 mutation) is more frequent in high-grade leukoplakias (severe dysplasia) (Lui et al., 1999). Accumulation of the mutated p53 in malignant oral epithelium has been demonstrated by several immunohistochemical studies (Zariwala et al., 1994; Saito et al., 1999). Elevated p53 in oral cancers correlates with heavy smoking but not with patient age (Field et al., 1991; Brennan et al., 1995b; Regezi et al., 1999). However, younger patients demonstrate p53 accumulation earlier during malignant progression (Castle et al., 1999). Mutant p53 has been investigated as both a diagnostic and a therapeutic adjunct (Brennan et al., 1995a; Breau and Clayman, 1996; Clayman et al., 1998, 1999). Last, p53 has been used as a prognostic marker for current head and neck cancer therapy (Dijkema et al., 2000; Obata et al., 2000; Temam et al., 2000; Warnakulasuriya et al., 2000). These studies mostly correlate the mutation of p53 with an unfavorable response to chemotherapy and radiation. However, the heterogeneity of p53 mutations renders the design of simple assays for this biomarker exceedingly difficult (van Houten et al., 2000).
(IV) Novel Cell Cycle Regulators
The signaling networks providing input into the cell cycle clock are continually being refined. While assigning cell cycle regulators to either extracellularly or intracellularly triggered pathways might help us begin to understand their biologic context, these designations are highly artificial: We are still discovering the pleotropic nature of the cell cycle regulators, as well as the overlap and interplay of the pathways where they function.
(A) p 12DOC-1
cell cycle biology
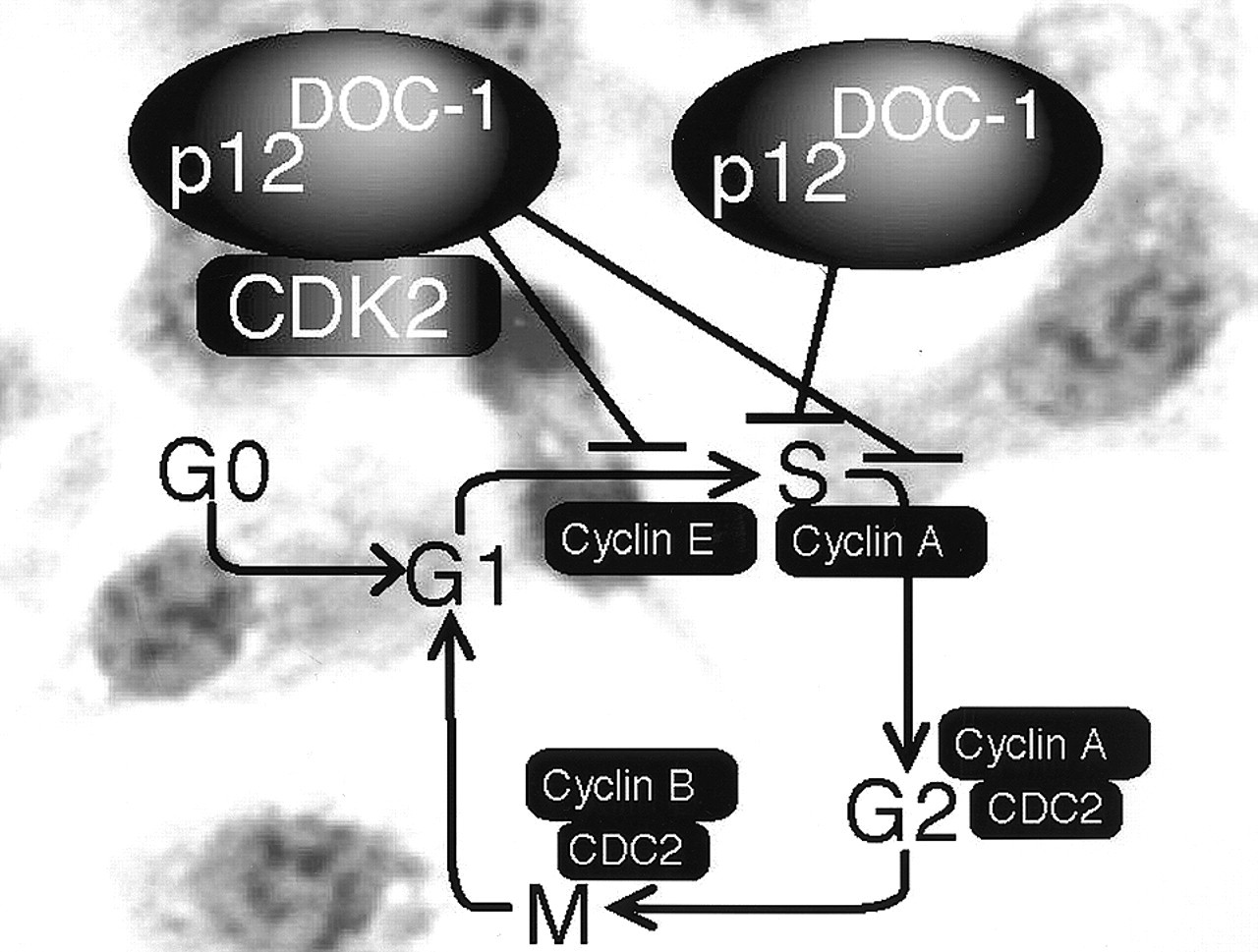
p12DOC-1, a growth suppressor identified and isolated from normal keratinocytes, has been identified to be a novel regulator of the cell division cycle (Todd et al., 1995) (Fig. 5). DOC-1 is a highly conserved cellular gene located on chromosome 12q24 whose cDNA has been cloned from humans, mice, and hamsters (Gordon et al., 1992; Todd et al., 1995; Daigo et al., 1997; Tsuji et al., 1998). Homologues have been located in EST databases of rat, Drosophila, and C. elegans. Human p12DOC-1 is a 115-amino-acid peptide with a molecular mass of 12.4 kDa (pI of 9.62) and with a 97% amino acid identity with the mouse and hamster p12DOC-1 protein sequences.
The cell-cycle-related function of p12DOC-1 was recently revealed by two sets of parallel studies. p12DOC-1 was found to interact with two cellular proteins of importance in S phase biology: DNA polymerase-α/primase (Matsuo et al., 2000) and CDK2 (Shintani et al., 2001a). The pol-α:primase binding domain in p12DOC-1 is mapped to the amino terminal six amino acid (MSYKPN). The biological effect of p12DOC-1 on pol-α:primase was examined by means of in vitro DNA replication assays. With the SV40 DNA replication assay, p12DOC-1 was shown to suppress DNA replication, leveling at about 50%. Similar results were obtained with the M13 single-stranded DNA synthesis assay. Analysis of the DNA replication products revealed that p12DOC-1 affects the initiation step, not the elongation phase. The p12DOC-1 suppression of DNA replication is likely to be mediated either by a direct inhibitory effect on pol-α:primase or by its effect on CDK2, a recently identified p12DOC-1-associated protein known to stimulate DNA replication by phosphorylating pol-α:primase. p12DOC-1 suppresses CDK2-mediated phosphorylation of pol-α:primase. Analysis of these data supports a role of p12DOC-1 as a regulator of DNA replication by direct inhibition of pol-α:primase or by negatively regulating the CDK2-mediated phosphorylation of pol-α:primase. Pol-α:primase is the only eukaryotic enzyme that can initiate DNA replication de novo. While p21WAF1/CIP1 has been shown to associate with proliferating-cell nuclear antigen (PCNA) and to inhibit DNA replication (Waga et al., 1994), analysis of our data demonstrates a proximal control mechanism whereby the first step of DNA replication, initiation, can be regulated upon interaction with p12DOC-1, suggesting that p12DOC-1 is a regulator of DNA replication negatively regulating the activity of pol-α:primase.
p12DOC-1 has also been found to associate with CDK2. More specifically, p12DOC-1 associates with the monomeric non-phosphorylated form of CDK2 (p34CDK2). Ectopic expression of p12DOC-1 resulted in decreased cellular CDK2, reduced CDK2-associated kinase activities, and a shift in the cell cycle positions of p12DOC-1 transfectants (↑ G1 and ↓ S). The p12DOC-1-mediated decrease of CDK2 is prevented if the p12DOC-1 transfectants were grown in the presence of the proteosome inhibitor clasto-lactacystin β-lactone, suggesting that p12DOC-1 may target CDK2 for proteolysis. A CDK2 binding mutant was created and found to block p12DOC-1/CDK2-associated cell cycle phenotypes. Analysis of these data supports p12DOC-1 as a specific CDK2-associated protein that negatively regulates CDK2 activities by sequestering the monomeric pool of CDK2 and/or targets CDK2 for proteolysis, thereby reducing the active pool of CDK2.
Expression of p12DOC-1 is cell-cycle-dependent and peaks in the S phase. The effect of p12DOC-1 on the biochemical pathway of CDK2 activation does not affect downstream phosphorylations by WEE1 and CAK. WEE1 and CAK kinase activities are not affected in the presence of p12DOC-1. The net effect of the interaction of p12DOC-1 with CDK2 is therefore the reduction of the intracellular pool of active CDK2, leading to the suppression of CDK2-mediated cell cycle phenotypes (G1/S transition, S phase progression, and DNA replication). CDK2-mediated cell cycle phenotypes are suppressed in cells ectopically expressing p12DOC-1. It is consistently observed that when cells are transfected with p12DOC-1, there is an associated growth suppression, change in cell cycle profile (↑G1 and ↓S), and suppression of DNA replication (p < 0.05) (Shintani et al., 2001a). It is intriguing to note that while the p21WAF1/CIP1 family of CDK inhibitors is universal for CDKs and the p16INK4a family is specific for CDK4 and CDK6, p12DOC-1 may be a specific CDK2-associated protein that suppresses CDK2 activities.
The role of p12DOC-1 in oral cancer cell cycle dysregulation has yet to be fully elucidated. In surveys including rodent oral keratinocyte cell lines, expression of DOC-1 has been consistently found to be down-regulated or lost in malignant keratinocytes (Todd et al., 1995), human cell lines, and oral cancer tissues (Tsuji et al., 1998). Reduced expression of p12DOC-1 in human oral cancer tissues correlated with negative prognostic indicators such as tumor mode of invasion (p = 0.0314), risk of cervical lymph node metastasis (p = 0.0226), and reduced patient 10-year survival status (p = 0.0032) (Shintani et al., 2001b).
(V) Cell Cycle Dysregulation and Multistep Carcinogenesis
Malignant progression is a multistep process. Escape of the cancer cell from cell cycle regulation reflects an important aspect of this multistep process (Lundberg and Weinberg, 1999). In many ways, pre-malignant cells with activated oncogenes or inactivated tumor suppressor genes are frequently prevented from clonally expanding by internal sensing mechanisms that lead to the induction of senescence (growth arrest) or apoptosis. Activation of the MYC oncogene results in the induction of apoptosis, while RAS activation induces senescence (Evan et al., 1992; Serrano et al., 1997). Therefore, the pathways leading to senescence or apoptosis must be abrogated in pre-malignant cells for malignant progression to occur. Alteration of p16INK4a activity and the association of human papillomavirus (HPV) during oral carcinogenesis are examples of the importance that cell cycle abrogation can be coupled with escape from differentiation, senescence, or apoptotic programs during malignant progression (Goodger et al., 1997).
(A) p 16INK4a
and multistep carcinogenesis
Dysregulation of the cell cycle regulators, such as p16INK4a, not only leads to uncontrolled cellular proliferation, but also contributes to escape from other checkpoints such as senescence and apoptosis. Inactivation of the CDKN2A locus occurs in the majority of oral cancers (Wu et al., 1999). Loss of p16INK4a inhibition of cyclin D1/CDK4 inhibition of the G1->S transition is a major abrogation of cell cycle regulation. p16INK4a activity leads to replicative senescence, and this phenotype can be restored in oral malignant oral keratinocytes that have ceased to express p16INK4a due to promoter methylation by 5-aza-2' deoxycytidine treatment (Timmermann et al., 1997). In addition, an alternative reading frame of p16INK4a encodes the p19ARF protein (Quelle et al., 1995). p19ARF inhibits p53 degradation by antagonizing MDM2 (Haupt et al., 1997; Kamijo et al., 1998; Zhang et al., 1998). Alteration of MDM2 expression has been associated with the pre-invasive stages of oral carcinogenesis and may suggest a more biologically aggressive tumor (Agarwal et al., 1999; Partridge et al., 1999). p53 can lead to either growth arrest or apoptosis, depending on the severity of the insult and the individual cell type response. Several reports implicate the p53 pathway as an important mediator of apoptosis in oral keratinocytes (Lui et al., 1999; Schoelch et al., 1999). In addition, loss of p53 activity also reduces p21WAF1/CIP1 induction of senescence (Ng et al., 1999). Once allowed to expand clonally, the pre-malignant cells continue to grow and replicate their genome regardless of unrepaired damage (Lin et al., 1991; Livingstone et al., 1992). Therefore, the likelihood of one of the pre-malignant clones undergoing the next critical genetic 'hit' increases markedly. Coupling cell cycle dysregulation with the abrogation of differentiation, senescence, and apoptotic programs may also be virally mediated during oral carcinogenesis.
(B) Human papillomaviruses and oral cancer
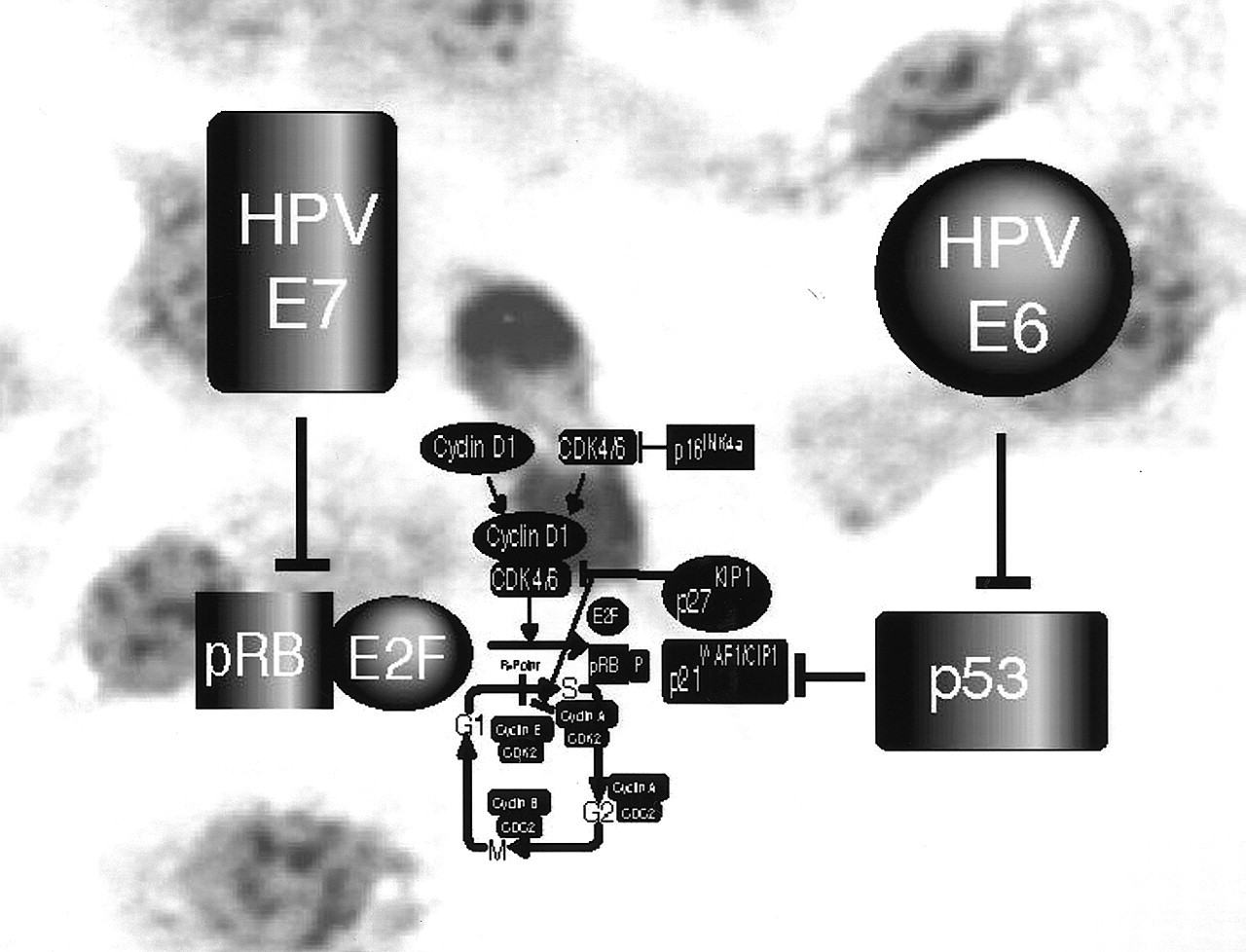
Infections of the genital tract epithelia with specific HPVs can lead to a variety of clinical outcomes. Low-risk HPVs cause benign genital warts such as condyloma acuminata, which only rarely progress to carcinoma. In contrast, however, infection with a high-risk HPV causes pre-malignant squamous intraepithelial lesions (SILs), which in some cases can progress to invasive cervical carcinoma. More than 90% of all cervical carcinomas express high-risk HPV. Since the high-risk HPV E6 and E7 oncoproteins, which are consistently expressed in these cancers, can inactivate the p53 and pRB tumor suppressor pathways, a high-risk HPV infection can functionally substitute for at least two mutational hits and mechanistically contribute to the multistep process of epithelial carcinogenesis (zur Hausen, 1996) (Fig. 6).
In the cervix, most HPV infections occur at the transitional zone between squamous and columnar epithelium (the 'transformation zone'), presumably because replicating basal epithelial cells are most easily accessible to the virus at this location. A similar junction of stratified squamous and columnar epithelium exists between the esophagus and the stomach. This anatomical location is much less accessible to HPV transmission than the cervical transitional zone. Thus, in a healthy individual, HPV infection of basal cells may be a rare event in the oral cavity. Severe alcohol and tobacco abuse and poor oral hygiene lead to ulcerations and other tissue damage in the oral cavity, and these changes are significant additional risk factors for oral carcinogenesis (Field, 1992). Similar to anogenital keratinocytes, primary cultures of oral keratinocytes are immortalized by high-risk HPVs (Park et al., 1991), and these cell lines progress to a fully transformed phenotype when treated with carcinogens (Kim et al., 1993), coinciding with increased genomic instability (Shin et al., 1996). This observation indicates that the biochemical activities of the E6/E7 oncoproteins have the same biological consequences in epithelial cells of both oral and anogenital origin.
Studies addressing the involvement of HPV in oral carcinogenesis have yielded ambiguous results (Yeudall, 1992). Although high-risk HPVs are detected in oral cancers, their possible role in oral carcinogenesis is obscure. Some studies have indicated that oral cancers may contain sequence variants of known high-risk HPV types (Maitland et al., 1987; Yeudall and Campo, 1991). In particular, it has been reported that oral-cancer-associated HPV variants may have mutations in the long control region that result in increased expression of the E6 and E7 genes compared with the prototypical HPV isolate (Chen et al., 1997). A very recent study demonstrated that about 25% of oropharyngeal squamous cell carcinomas are positive for integrated HPV genomes (Gillison et al., 2000). This finding suggests that HPV-positive head and neck cancers may constitute a distinct class of disease with better prognosis than those that are HPV-negative. These tumors can occur in patients without other risk factors (smoking, alcohol consumption) and are less frequently associated with p53 mutations than HPV-negative cancers.
(VI) Future Directions and Clinical Applications
The oral cancer problem primarily involves the understanding, diagnosis, and treatment of squamous cell carcinoma of the oral cavity (Silverman, 1988). While recent advances have reduced the morbidity of oral cancer, the five-year survival rate for these patients has remained largely unchanged at ~ 50% for the last 30 years, because early stages of the disease are associated with minimal signs and symptoms, and advanced stages generally respond poorly to current cancer therapies (Silverman, 1998). This review highlights the current status of investigative efforts to understand the identity and function of specific cell cycle defects in human oral cancer. While the majority of oral cancers harbor p53 mutations, CDK6 hyperactivation is a unique defect in the pRB tumor suppressor pathway, in addition to frequent cyclin D1 overexpression and p16INK4a mutations. The ability to map the signature cell cycle defects in human oral cancer is of value not only for the biological understanding of the disease but more importantly toward the translational utilization of this information for early diagnosis and biology-based therapy. Many of the cell cycle regulators reviewed have been associated as biologic predictors of oral cancer behavior. In the future, our ability to profile, comprehensively, the gene expression differences among normal, pre-malignant, and tumor cells from the same patient will allow us better to index the consistently altered cell cycle defects in human oral cancer (Shillitoe et al., 2000). Development of new animal models will advance our understanding of the functional consequences of these cell cycle defects. Understanding the identity and function of oral cancer cell cycle defects will provide novel biological/genomic parameters for patient outcome monitoring and treatment therapy decisions and options.
Two new head and neck cancer therapeutic approaches under investigation are cell-cycle-based. ONYX-015 is an E1B attenuated adenovirus that is believed to replicate selectively in p53 mutant cells, therefore sparing normal/wild-type p53 cells (Bischoff et al., 1996). However, other studies fail to correlate mutant p53 and viral replication (Hall et al., 1998). When administered intratumorally to patients with recurrent head and neck cancer, ONYX-015 produced tumor necrosis in four of five patients with mutant p53 (Ganly et al., 2000). ONYX-015 treatments, which were easily administered and well-tolerated, may serve as an adjunct to current chemotherapeutic approaches to head and neck cancer patients (Kirn et al., 1999). Recently, a phase II trial on recurrent head and neck cancers was completed, demonstrating improved response to intratumoral ONYX-015 injection with cisplatin and 5-fluorouracil vs. either viral treatment or chemotherapy alone (Khuri et al., 2000). Flavopiridol is a novel cyclin-dependent kinase inhibitor that has been demonstrated to have anti-neoplastic properties (Weinstein et al., 1997). Recently, flavopiridol has been shown to suppress head and neck carcinoma growth by inducing apoptosis (Patel et al., 1998). Exposure of malignant oral keratinocytes to flavopiridol diminished CDC2 and CDK2 activity, as well as reduced cyclin D1 expression. Certainly, understanding the mechanisms of cell cycle dysregulation of the oral keratinocyte during oral carcinogenesis will serve as an adjunct to present diagnostic and therapeutic options and provide the basis for novel strategies in the future.
Regulation of the G1 to S phase transition by the retinoblastoma suppressor protein (pRB). G0, G1, S, G2, and M refer to the quiescence, first gap, DNA synthesis, second gap, and mitosis phases of the cell cycle. CDK refers to cyclin-dependent kinases. CDC-2 refers to cell cycle control-2. Phosphorylated pRB is represented as pRB-P. Briefly, pRB-E2F transcriptional repression of genes regulating DNA synthesis is released by phosphorylation of pRB, allowing for cell cycle progression from G1 to the S phase. Mitogen-activated G1 transition to the S phase. Mitogen stimulation leads to cyclin D1 synthesis. Together with its catalytic partners CDK4 and CDK6, cyclin D1 accelerates G1 progression by phosphorylating pRB. Cell cycle block by anti-mitogenic signals. Anti-mitogenic signals stimulate cyclin-dependent kinase inhibitors (CDKi) such as p16INK4a, p21WAF1/CIP1, and p27KIP1. Conversely, mitogenic stimulation leads to reduced levels and loss of CDKi-mediated inhibition of cyclin D1/CDK complexes. p53 and cell cycle regulation. Metabolic stresses (such as anoxia) and DNA damage lead to elevated p53 activity, resulting in cell cycle arrest and apoptosis. p12DOC-1-mediated cell cycle arrest. p12DOC-1 is a CDK2-associating protein, binds to the monomeric non-phosphorylated form, and suppresses the binding to cyclin E and cyclin A. In addition, p12DOC-1 suppresses DNA replication by binding DNA polymerase-alpha/primase. Multistep abrogation of cell cycle regulation. Dysregulation of cell cycle progression likely requires both a direct regulatory checkpoint loss combined with a failure to activate cellular senescence, differentiation, and/or apoptotic programs. The human papillomavirus (HPV) synthesizes two proteins, E6 and E7, that abrogate the p53 and pRB pathways.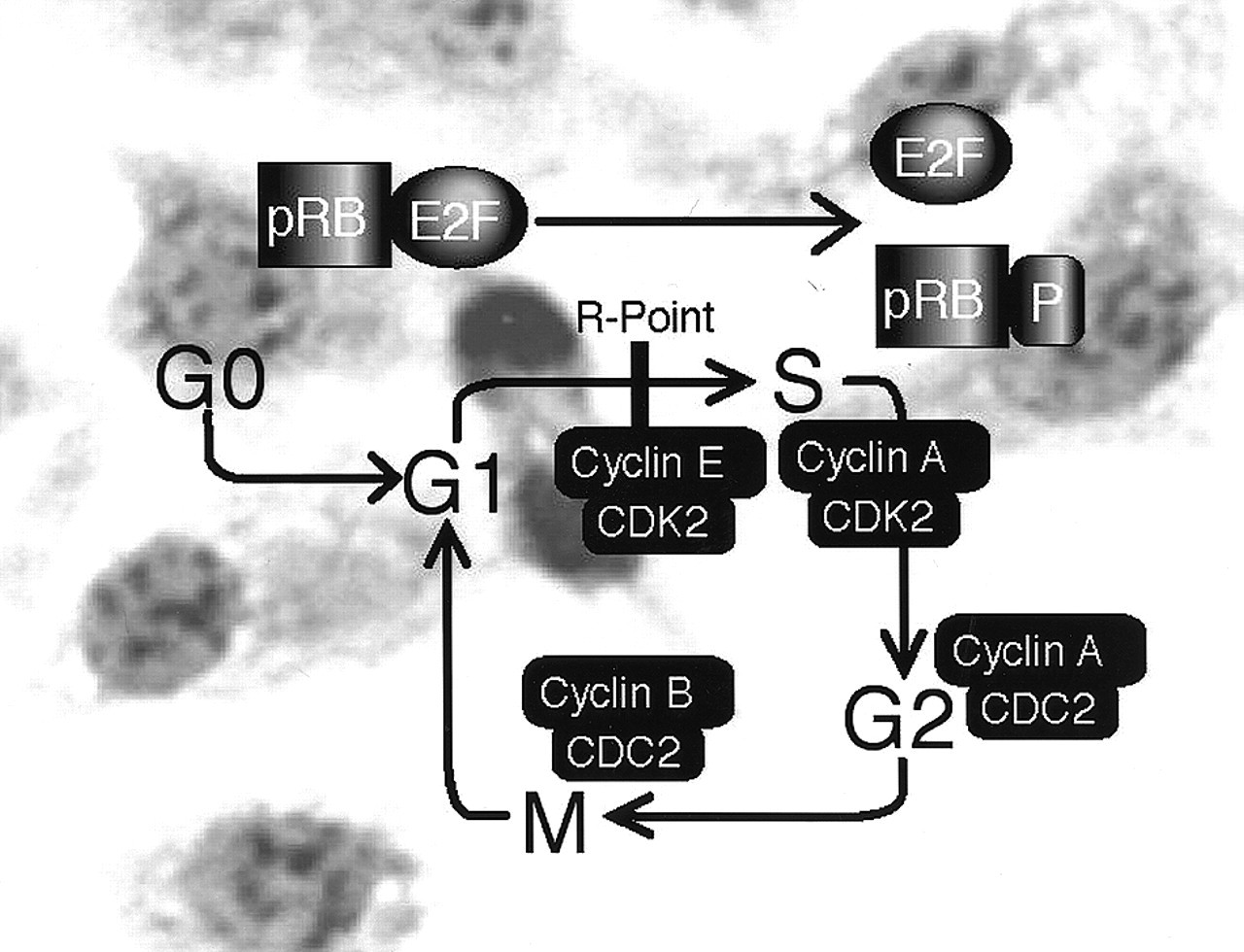
Footnotes
Acknowledgements
Supported by the National Institute of Dental and Craniofacial Research (P01 DE12467).