
Abstract
Despite the significant progress made in developing different in silico methodology for structure activity research over the past few decades. The ability to predict correlation structure activity (CSA) from absorption distribution metabolism excretion (ADME) descriptors to select indolizine compounds for human papilloma virus (HPV) anticancer activity continues to pose a challenge. This study employed five machine learning (ML) algorithms for classification, viz., stochastic gradient descent (SGD), random forest (RF), support vector machine (SVM), convolutional neural network (CNN), and logistic regression (LR), to perform the classification based on ADME-related physiochemical descriptors of 8,900 indolizine compounds to predict the CSA. The present study focuses on 26 well-known parameters to optimize the results, which are utilized for ML models SGD, RF, SVM, CNN, and LR for classification. The CNN achieved the best results with the highest overall accuracy and average loss values of 98.33% and 0.16, respectively. On the other hand, the SGD, RF, SVM, and LR recorded the accuracy values of 95.32%, 93.23%, 96.03%, 94.03%, and loss values of 0.046, 0.067, 0.039, and 0.059, respectively. It is stated that from the obtained results, the CNN is performing better compared to other methods. The cross-validation and results are done with the relationship of descriptors, viz., accuracy, correlation, distribution, area under the receiver operating characteristic, area under the precision recall curve, and bootstrap error analysis. This study demonstrated the utility of ML to facilitate early prediction of indolizine compounds for HPV anticancer activity in preclinical development.
This is a visual representation of the abstract.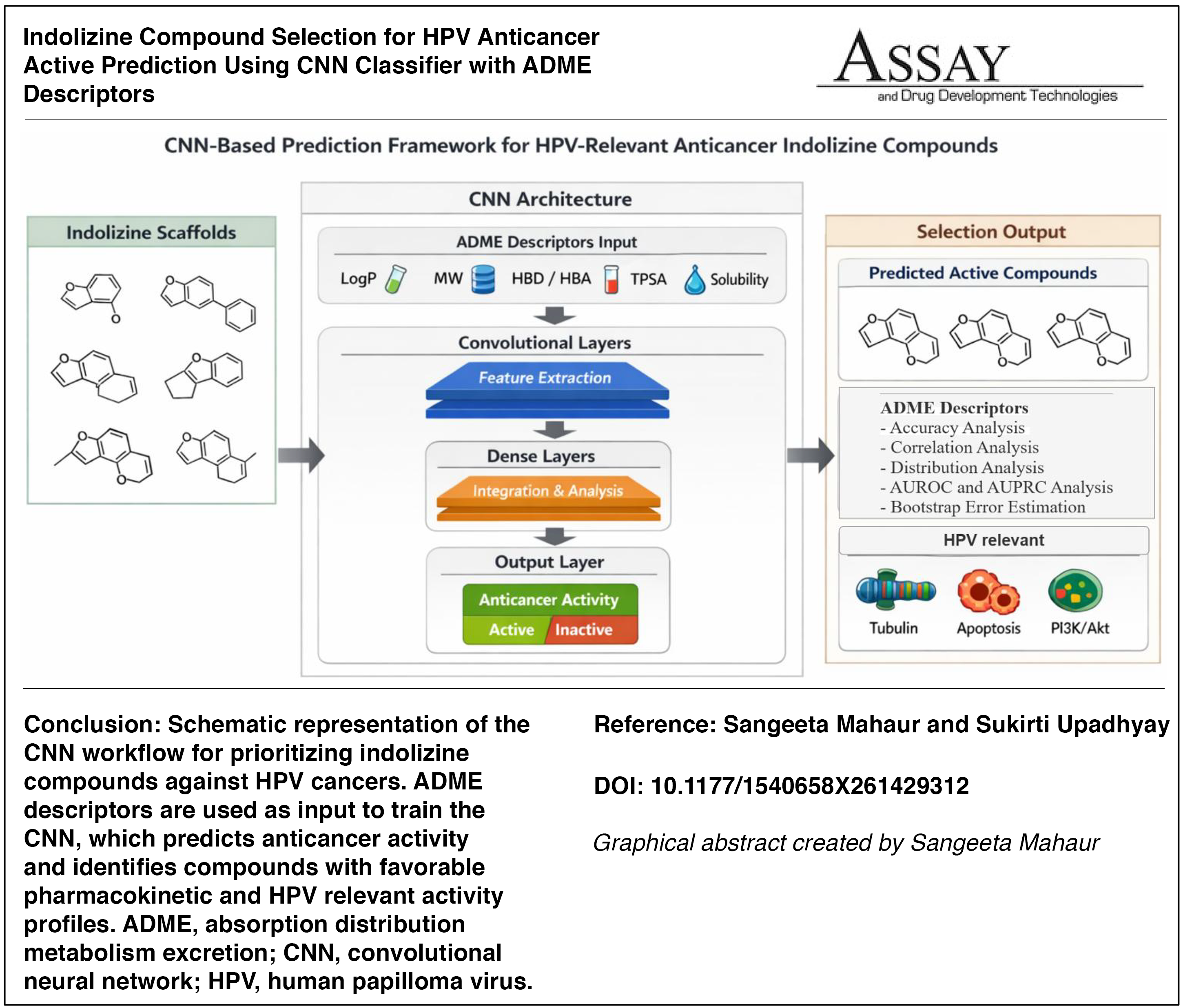
Get full access to this article
View all access options for this article.