
Abstract
Hydrolytic enzymes are a large class of biological catalysts that play a vital role in a plethora of critical biochemical processes required to maintain human health. However, the expression and/or activity of these important enzymes can change in many different diseases and therefore represent exciting targets for the development of positron emission tomography (PET) and single-photon emission computed tomography (SPECT) radiotracers. This review focuses on recently reported radiolabeled substrates, reversible inhibitors, and irreversible inhibitors investigated as PET and SPECT tracers for imaging hydrolytic enzymes. By learning from the most successful examples of tracer development for hydrolytic enzymes, it appears that an early focus on careful enzyme kinetics and cell-based studies are key factors for identifying potentially useful new molecular imaging agents.
Keywords
PET and SPECT Imaging
Positron emission tomography (PET) and single-photon emission computed tomography (SPECT) are nuclear-based molecular imaging modalities. They are used for noninvasively tracking an injected molecule labeled with a radioactive isotope to study its spatial and temporal distribution in a living subject to gain fundamental information about disease biology. 1 Today, PET-CT and SPECT-CT instruments are both available as dual imaging systems enabling anatomical computed tomography (3-D X-ray) images to be overlaid with the radioactive signal. Such techniques enable real-time imaging of physiological processes such as blood flow and cardiac function, but they truly excel at “molecular imaging” of unique disease-specific metabolic processes including the activation of enzymatic pathways and cellular processes such as receptor expression, angiogenesis, and apoptosis. Positron emission tomography and SPECT are used extensively in both preclinical and clinical settings due to their high sensitivity and total body penetrance, which enable detection of the tracer in any tissues or organs in the full body. With careful design and selection of the radiotracer, these scans can serve as the equivalent of a noninvasive whole-body biopsy for a given process, as the nature of nuclear imaging scans allows quantitative measurement of radiotracer distribution. Owing to the high sensitivity of PET and SPECT, only microdoses of radiotracer are needed (final biological concentrations typically nM-pM), which generally avoids unwanted pharmacological side effects of the chemical probe. However, the design of PET and SPECT tracers capable of imaging enzymatic targets is an enormous challenge due to the need for agents that have high affinity and specificity toward the target enzyme, possess good clearance properties to reduce background signal from nonspecific tissue uptake, and are metabolically stable. In addition, the preparation, purification, and quality control of radiotracers are demanding due largely to the short half-life of many commonly used isotopes (especially for PET). Other challenges include poor penetration of the tracer into specific tissues such as tumors or organs such as the brain, limitations associated with using animal models as a surrogate to human diseases, and the difficulty and costs of obtaining sufficient data for Food and Drug Administration approval. 2 The versatile functionality and general success of nuclear imaging and new radiopharmaceuticals have led to increasing availability of PET and SPECT cameras at many research centers and urban hospitals, further increasing the need for new and innovative molecular imaging agents.
Positron emission tomography utilizes isotopes with an energetically unstable proton to neutron ratio (eg, 11C, 18F, 68Ga, 64Cu, 89Zr), which are often produced in a cyclotron or a generator system, where the generator parent isotope may be produced via cyclotron or nuclear reactor. These radioisotopes decay through emission of a positron (the antimatter equivalent of an electron bearing a positive charge) that travels a short distance before colliding with an electron. Mutual annihilation of both particles results in the release of 2 coincident 511 keV gamma rays, which are detected simultaneously by a PET camera (coincidence detectors).
For PET imaging, 18F has traditionally been the isotope of choice because of its 109.7-minute half-life, ready availability at virtually every medical cyclotron facility in the world, and good image resolution as a result of low positron energy. Compared to 11C with its 20-minute half-life, the longer lived 18F isotope allows for the preparation of a wider range of radiotracers (with well-established radiolabeling chemistry) and provides more working time for purification, quality control, and distribution to nearby hospital radiopharmacies while still possessing a short enough half-life to minimize radiation dose to patients. Another attractive property of 18F compared to the radiometals is that it can be seamlessly substituted into small organic molecules as the change from a C-H or C-OH to a C-F bond is relatively conservative in terms of bond length, bond strength, and atomic radius. Although 18F is a near-perfect match for labeling small molecules intended for imaging, the use of radiometals has seen a surge of interest with larger drug vectors such as peptides, antibodies, and nanoparticles. 3
As a consequence of their short half-lives, PET isotopes such as 11C and 18F require production at a nearby cyclotron facility, which significantly increases the costs associated with PET imaging. Long half-life isotopes for PET (eg, 89Zr, 124I) and SPECT (eg, 111In, 123I, 131I) are available but are not always as structurally compatible with small molecule drugs as either 11C or 18F due to their large size or need for chelation (eg, 18F is bioisosteric with hydrogen, bulky chelators are not).
Compared to SPECT, PET has higher sensitivity and therefore requires preparation and injection of a lower dose of radiotracer, is quantitative for more accurate pretherapy scouting scans and dosimetry calculations, and has fewer image artifacts leading to better quality images. Positron emission tomography is generally considered a superior technique for molecular imaging. In contrast, SPECT isotopes are longer lived (eg, 99mTc t 1/2 = 6.00 hours; 123I t 1/2 = 13.2 hours, 111In t 1/2 = 2.8 days) and decay through the direct emission of single gamma ray photons that are detected directly with pinhole cameras through collimators, which greatly reduces sensitivity when compared to noncollimated coincidence detection of PET. With SPECT cameras located in virtually every nuclear medicine department worldwide, SPECT represents ∼90% of all nuclide-based diagnostic medicine despite having inferior image quality and lower sensitivity compared to PET. Among the available SPECT isotopes, 99mTc is the most commonly used (∼75% of all nuclear diagnostic tests) due to the low cost of reactor-produced 99Mo and the small and easily distributed 99Mo/99mTc generator system. These generators are purchased by hospitals and research centers where the parent isotope 99Mo remains trapped at the top of the generators’ stationary phase, continually decaying into a supply of 99mTc. The 99mTc can be eluted (referred to as “milking”) daily for about a week and used directly with radiopharmaceutical kits for rapid production of the SPECT radiotracer. A majority of the world’s 99Mo supply is produced from highly enriched uranium in a handful of nuclear reactors: the National Research Universal reactor in Canada, High Flux Reactor in the Netherlands, Osiris in France, Belgium Reactor-2 (BR-2) in Belgium, and South Africa Fundamental Atomic Research Installation (SAFARI-1) in South Africa. As well, low-enrichment uranium is being used to produce 99Mo at a small number of reactors, including Australia’s Open Pool Australian Lightwater (OPAL) reactor. 4,5 Despite its wide availability, 99mTc requires most radiotracers to incorporate a bulky chelating group and is therefore not commonly used to label small molecules intended to image enzyme activity.
Imaging Hydrolytic Enzymes
Hydrolases (abbreviated Enzyme Commission number (EC) 3 in the International Union of Biochemistry and Molecular Biology Enzyme Classification System) are a large family of enzymes responsible for breaking down biological molecules through addition of water followed by fragmentation of the substrate. 6 Hydrolytic enzymes have a variety of critical roles in normal cell and tissue function but are also important in a large number of disease processes, including viral, parasitic, or bacterial infection, cancer, neurodegenerative diseases, and addiction. Tremendous efforts have been made in developing radiotracers to image in vivo hydrolase activity in animals and humans, with the goals of identifying enzyme-based biomarkers for diagnosing disease, understanding the role that a specific enzyme may play in disease-promoting processes, or identifying aberrant enzyme activity as a putative drug target. Indeed, many PET/SPECT agents have been developed to image esterases (EC 3.1), glycosylases (EC 3.2), proteases (EC 3.4), other carbon–nitrogen cleaving enzymes (EC 3.5), and, to our knowledge in only 1 case, ether hydrolases (EC 3.3). This review is focused on describing examples and highlighting the various strategies recently published in the scientific literature to image hydrolase enzyme activity using molecular imaging. In the case of hydrolytic enzymes recently reviewed in a comprehensive manner, an interested reader is directed to relevant articles without further comment. Furthermore, enzymes with hydrolytic activity that is secondary to the primary function of the protein, for example, an enzyme that couples adenosine diphosphate hydrolysis to another reaction, have been excluded.
General Considerations for the Design of Enzyme Imaging Agents
The design and development of PET/SPECT tracers that can image enzyme activity can be classified into 3 broad categories, described below.
Substrates
A key design feature of a substrate-based radiotracer for a hydrolase is that the enzyme activation of the radiolabeled substrate creates 2 fragments. One of the product fragments produced must both retain the radioactive label (eg, 18F) and become trapped in nearby cells or tissues so that the accumulation of the radioactive signal correlates with areas of high enzymatic processing (and high enzymatic activity). It is important to note that radioactive PET/SPECT images do not provide any information about the chemical state of the isotopes (eg, bound to tracer or not), and for example, isotope that is “lost” from the parent radioactive drug (eg, 18F being released, or on the wrong side of a hydrolyzed substrate) is indistinguishable from tracer-bound isotope. One of the major challenges in design of a substrate-based imaging agent is selectivity for the target enzyme. Designing a substrate-based tracer that will only reflect the activity of a single enzyme is challenging since closely related enzymes (isozymes) often recognize similar substrates and have therefore evolved very similar active site structures comprised of conserved amino acid residues. As well, a substrate must be processed with very high efficiency so that even at tracer levels of the substrate, sufficient probe is hydrolyzed and enough radioactive products accumulate to enable detection of enzyme activity prior to elimination or extensive decay of the radioisotope (eg, 1-4 hours postinjection for 18F). A major hurdle to overcome in the substrate-based approach is in the selection of the radiolabeled fragment that must undergo cellular or metabolic trapping: It must be easily radiolabeled, compatible with the enzyme active site, and reliably accumulate at the site of enzyme activity. Common strategies developed for trapping radiolabeled reporter groups include precipitation of hydrophobic products, intracellular accumulation of charged products, and reaction to form a covalent bond with a cellular component. 7 -10 Although not discussed here, substrate-based probes designed to image kinases have seen tremendous success because the phosphorylated product becomes ionically trapped inside cells (an ideal example of this approach is the phosphorylation of [18F]2-fluoro-2-deoxyglucose ([18F]FDG) by hexokinase). 11,12 Despite these challenges, substrate-based radiotracers have the very important advantage of signal amplification. An individual enzyme molecule can turn over many probe molecules, leading to a high accumulation of radioactive signal in the region of enzyme activity. As well, in many disease processes, aberrant enzyme catalytic activity is observed and is critical for progression. Given that human enzyme activity is tightly controlled through complex posttranslational modifications and not expression levels alone, substrate-based radiotracers can be used with noninvasive PET imaging to reveal only those enzymes that are catalytically active.
Reversible Inhibitors
This class of enzyme imaging tracer relies on the development of a potent radioactive ligand that noncovalently binds to a target with very high affinity (generally nM Ki ). Since the radioactive ligand will bind to the desired enzyme in a reversible manner, the PET/SPECT tracer will accumulate in areas with high levels of target enzyme leading to a strong radioactive signal. In tissues with low enzyme concentration, the tracer can diffuse away and is eliminated from the body (eg, urine/renal clearance), effectively reducing background uptake and improving image contrast. Frequently, inhibitors designed as therapeutics can serve as the inspiration for the design of reversible inhibitor-based radiotracers. Indeed, some failed drug candidates suffering from undesirable side effects or rapid metabolism can be converted into excellent tracers once radiolabeled analogues can be prepared. As radiopharmaceuticals are administered in tiny quantities as “microdoses,” pharmacological toxicity is far less of a concern. Another advantage of this approach is that potent ligands can be designed to bind at “allosteric sites” on the target enzyme that have often evolved as a specific way of controlling metabolic rates through inhibiting key enzymes. Given that allosteric sites often bind natural ligands specific to a metabolic pathway, tracers that occupy a unique allosteric site may be highly selective to the target enzyme over closely related homologous enzymes. 13 Reversible ligands image enzyme expression rather than enzyme activity since the ligand binds in a 1:1 ratio with enzyme regardless of whether the enzyme is catalytically active in the local environment. 14,15
Irreversible Inhibitors
The third class of tracer molecule is irreversible inhibitors. These compounds are substrate analogues that, once activated by the target enzyme’s catalytic machinery, generate a reactive intermediate that covalently attaches to a nucleophilic amino acid residue typically in the enzyme’s active site. Enzyme activity is permanently inhibited either because a catalytic residue no longer functions as needed or because the inhibitor physically blocks the active site. Because these inhibitors rely on catalytic activity but also irreversibly tag the target enzyme, they image enzyme activity (as opposed to expression levels) with a 1:1 inhibitor to enzyme ratio. Since a stable enzyme–tracer complex forms, the radioactive signal is stably attached to the enzyme and therefore cannot diffuse away from area of enzyme activity. One feature of imaging agents based on irreversible inhibitors that is fundamentally different from substrate-based tracers is that a large number of substrate molecules can be processed by each individual enzyme which results in signal amplification over time, whereas irreversible inhibitors are limited to a 1:1 tracer to enzyme ratio.
A drawback of this approach is that the inhibitor efficiency must be carefully tuned since compounds that are too potent will instantaneously react with the target enzyme producing images that can reflect blood flow instead of areas of high enzyme activity. The behavior of potent irreversible inhibitors to image blood flow results from the rapid labeling of the target enzyme in highly vascularized tissues that occurs much faster than the rate of diffusion through tissues that have low enzyme expression. In this scenario, the limiting reagent is the radiotracer—meaning the inhibitor reacts with only a small proportion of the available enzyme molecules and imaging experiments are therefore unable to distinguish between areas of high or low enzyme activity. 14,15
Two enzymes whose activity and expression have been successfully imaged with all 3 approaches are monoamine oxidases A and B (MAO A/B). These flavin-dependent oxidoreductase (EC 1.4.3.4) enzymes have been very well studied by Fowler and coworkers, and a number of imaging agents that are (1) substrates, (2) reversible, and (3) irreversible inhibitors have been described. 15
It is also possible to image an enzyme using a radiolabeled monoclonal antibody. The benefits and challenges surrounding PET/SPECT imaging using antibodies are different from those encountered in small molecule development, including the higher potential specificity, longer times needed to accumulate specific signal in target tissues (imaging is usually feasible after 24-74 hours), possibility for development of an immune response to the imaging agent (eg, murine vs human antibodies), and inability for antibodies to natively cross cell membranes or cross the blood–brain barrier (BBB). Since the fundamental design of antibody-based radiotracers is different from imaging with small molecules, 16 -19 this review will focus only on small molecule radiotracers. For a more complete description of various design strategies for PET imaging probes, the interested reader should consult specific reviews on that topic. 8 -10
Enzyme Kinetics for Tracers
The measurable parameters in an in vitro enzyme kinetic experiment are also different for each of the 3 classes of imaging agent. Each type of imaging agent has unique kinetic properties that can be measured using experiments conducted in vitro with recombinant enzymes or cultured cells. For a good introduction to the basics of enzyme kinetics, see the study by Cook and Cleland. 20
Substrates
Substrates for hydrolytic enzymes can be treated using classic Michaelis-Menten kinetics. 21 The Michaelis-Menten equation describes a kinetic scheme in which a single substrate rapidly binds to an enzyme and is transformed into product in a single, irreversible step. A kinetic scheme representing this process is shown in Figure 1.

Kinetic scheme for Michaelis-Menten kinetics, in which E represents free enzyme, S represents free substrate, ES represents the enzyme–substrate complex, and P represents product.
The equation that describes this process for velocity of the reaction (v) at concentration S is:

In this equation, the important parameters are V max and Km . V max is the maximal rate of reaction and is dependent on enzyme concentration. Dividing V max by the total enzyme concentration yields the important parameter kcat , which represents the rate of the first committed chemical step of the reaction at saturating substrate concentrations. Km is the concentration of substrate needed to reach half the maximum rate (V max) and represents the affinity of the substrate for the Michaelis complex (ES). A low Km value indicates that the enzyme has a high affinity for the substrate, meaning that even at low concentrations of a radiotracer, appreciable levels of hydrolysis will be occurring. In contrast, a high Km value indicates the enzyme requires a high concentration of radiotracer to reach high levels of substrate turnover. Km , the Michaelis constant, is sometimes mistakenly conflated with the dissociation constant (Kd value). These values are not equivalent, since Kd = k −1/k 1, while Km = (k 2 + k −1/k 1), therefore as kcat (which is = k 2 in Michaelis-Menten kinetics) increases, the value of Km will progressively get larger than the Kd value. In the limiting case where kcat for a substrate approaches 0, then the value of Km approaches Kd and the small molecule is behaving as a potential inhibitor for the enzyme (discussed below). The ratio of kcat /Km is the enzymatic efficiency, representing the most valuable measurement for evaluating substrates intended as tracers for imaging enzyme activity. This ratio is a second-order rate constant for reaction of free enzyme and free substrate to form product and accounts for both the binding affinity of the substrate and the catalytic efficiency of the enzyme for the substrate. 22 A high ratio for kcat /Km indicates that a substrate will be processed efficiently by the enzyme even at physiological concentrations and tracer levels of the radioactive substrate, which is a critical parameter for successful radiotracers (Table 1).
Key Kinetic Values for the Discussion of Enzyme-Targeted Molecular Imaging Agents.

In practice, potential substrates designed as tracers for imaging enzyme activity should have kcat and Km values rather than % activity measured, since kcat and Km values are independent of enzyme and substrate concentration. Measurements of % activity may be a useful guide for evaluating a series of substrates to select the most efficient, but without a proper kinetic characterization, it will be impossible to tell whether increases in efficiency arise from faster processing of the substrate (increased kcat ) or improved binding (roughly reflected by Km ). Furthermore, reporting % activity makes it very difficult to directly compare substrate efficiencies for assays performed in different laboratories, and so standardized kcat and Km values are far more useful.
The ideal substrates used for PET/SPECT imaging should have high kcat values, indicating the enzyme efficiently processes the substrate, and low Km values, indicating the enzyme requires a low concentration of substrate for efficiently processing. It can be assumed that at tracer levels (nM-pM), the [Substrate]<<Km , meaning that measuring kcat or Km alone will not provide information on overall efficiency. The kcat /Km value is of the most value for evaluating substrates as potential imaging agents, since this second-order rate constant describes the rate of free enzyme and free substrate converting to product at low substrate concentrations and takes into account both substrate binding and turnover rates. 22 The challenge here is that many potential substrates cannot be easily assayed unless they happen to incorporate a reporter group, like a chromophore or fluorophore. In addition, sufficient levels of nonradioactive probe as well as access to recombinant enzyme or the development of a cell-based kinetic assay are needed for full kinetic characterization and represent a significant hurdle in many cases.
Reversible Inhibitors
Reversible inhibitors bind to the enzyme and interfere with catalytic turnover of substrate. Reversible inhibitors can be classified as competitive (increases the apparent Km for a substrate, indicating it requires higher substrate levels), uncompetitive (decreases the apparent kcat for a substrate, indicating a decrease in efficiency of substrate processing), or mixed (apparently changes both kcat and Km ) depending on their kinetic behavior. This kinetic behavior can be modeled as the inhibitor binding to different forms of the enzyme, as seen in Figure 2. 23 , 24
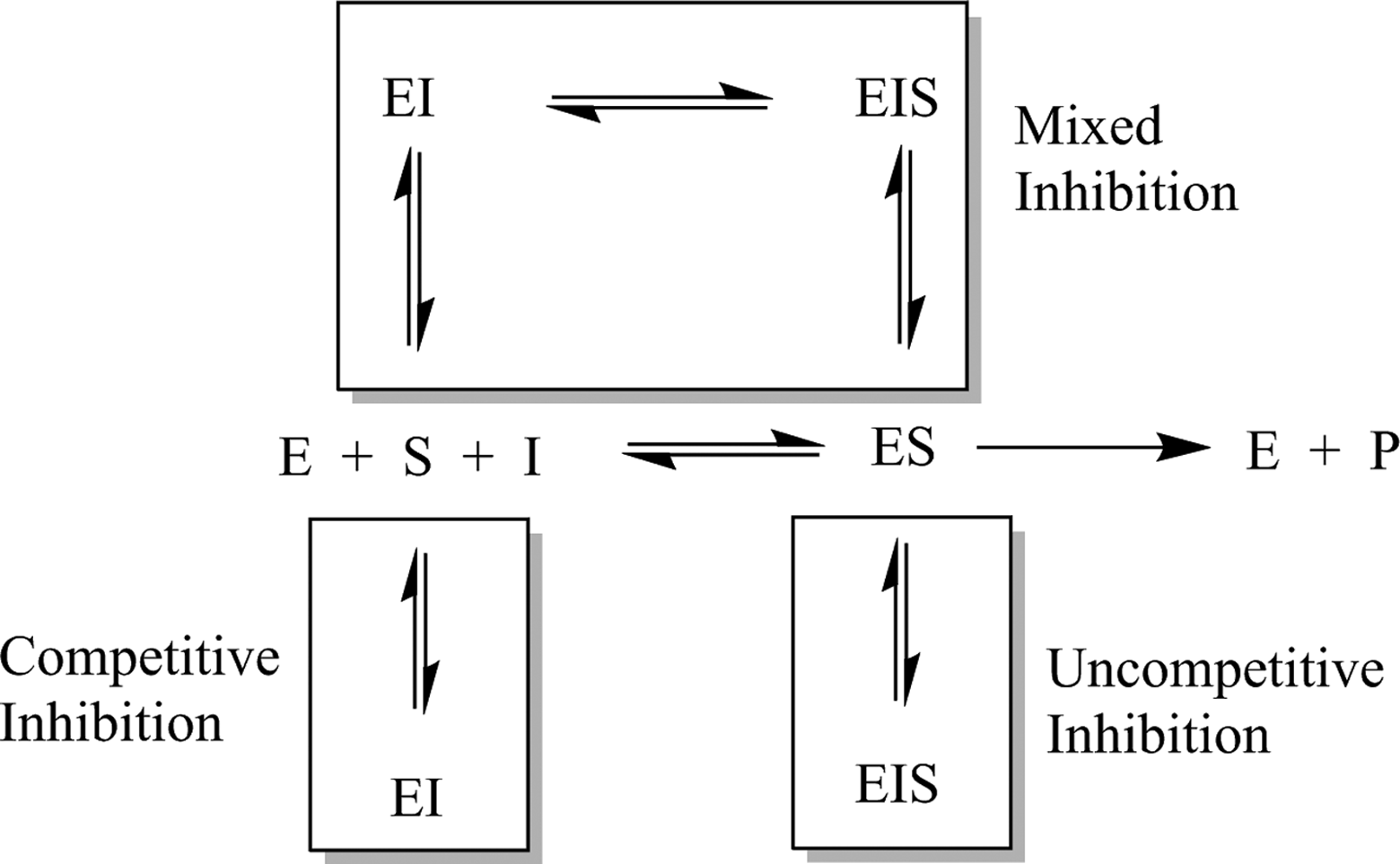
Kinetic scheme depicting enzyme-catalyzed turnover of substrate (center row) and various modes of reversible inhibition.
In practice, most inhibitors intended as nuclear imaging agents are competitive inhibitors, which can be conceptually thought of as binding to the active site and blocking substrate binding and subsequently catalysis. Therefore, substrate, product, or transition state analogues are often the starting point for inhibitor design. Reversible inhibitors should have either Ki or IC50 values measured. Ki is the equilibrium constant for binding of the inhibitor, which is the ratio of the rate constant for enzyme–inhibitor dissociation divided by the ratio of enzyme–inhibitor association (koff /kon ). IC50 is the concentration at which enzyme activity is reduced by 50% under given assay conditions. The advantage of measuring a Ki value is that it is independent of enzyme and substrate concentrations, so values measured in different laboratories, different cell lines, and under different conditions can be directly compared. As well, careful measurements of a Ki value will help identify and properly characterize potentially slow- and tight-binding reversible inhibitors. 25,26 Such incredibly potent inhibitors may have a biological effect even at tracer levels, similar to some of the most potent imaging agents for the opioid receptors. 27 The major advantage of measuring an IC50 value is that measurements are easier and faster (requiring only approximately 15%-20% of the number of data points 28 ) which can facilitate a quick determination of relative inhibitor potency when evaluating a number of potential inhibitors as imaging tracers or when quickly validating a known substrate that has been modified for molecular imaging (eg, radiolabeled peptides, drugs).
Ideal reversible inhibitors used as imaging agents should have low Ki or IC50 values, since those potent inhibitors will concentrate more readily in sites of enzyme expression. However, there is a practical lower limit for the Ki value (koff /kon ) of inhibitor intended for imaging enzyme distribution. Since kon is limited by the rate of diffusion (∼108 M−1s−1), 29 extremely strong inhibitors must derive their potency from reducing the rate of koff . In practice, this means that inhibitors with Ki < ∼10 nM are members of a subclass called tight-binding inhibitors 25,26 and will not rapidly equilibrate with their target enzyme. The resulting images will not reflect enzyme expression levels, because tracer washout does not occur due to slow off rates and therefore does not reach an equilibrium distribution as kinetic modeling for a reversible inhibitor demands. Indeed, a very potent inhibitor begins to behave kinetically more like an irreversible inhibitor (discussed below) rather than a reversible inhibitor leading to images that do not reflect differences in enzyme distribution within a given tissue or organ. 14 This information is needed for each individual radiotracer used for imaging different enzymes, as correct classification (eg, reversible vs irreversible vs tight-binding reversible) is needed to perform correct kinetic modeling and properly interpret PET imaging data. The dissociation of the tight-binding inhibitor methotrexate from dihydrofolate reductase demonstrates how slow off rates can affect the lifetime of the inhibitor–enzyme complex: The half-life for dissociation of the enzyme–inhibitor complex, as estimated by dialysis experiments, is greater than 6 days. 30 If such an inhibitor were to be radiolabeled and monitored using PET/SPECT, kinetically modeling assuming reversible inhibition (with rapid enzyme–inhibitor association–dissociation rates) would be clearly inappropriate.
Irreversible Inhibitors
Irreversible inhibitors reduce enzyme activity through formation of a stable covalent bond between the inhibitor and the target enzyme. These are most often mechanism-based inactivators, which are substrate analogues that are activated by the enzyme’s catalytic machinery to reveal a reactive moiety (most often a latent electrophile) that subsequently covalently binds to an amino acid residue in the active site resulting in a loss of activity. Inhibition can occur through covalent modification of a key catalytic residue (eg, alkylation of a carboxylate or amine sidechain) or by physically blocking access to the active site. Kinetically, this process is analogous to processing of a substrate by the enzyme and can be represented by Equation 2, which is described in Figure 3.
31

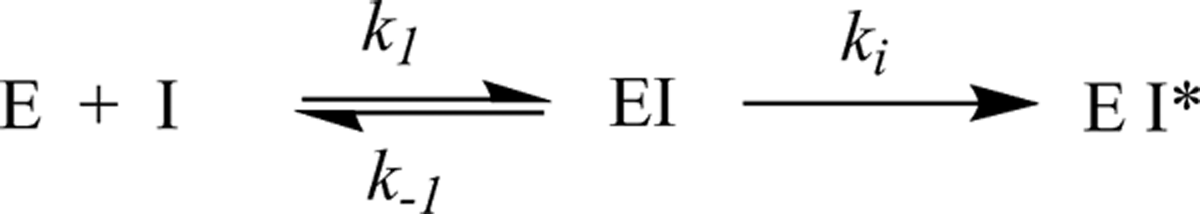
Kinetic scheme for irreversible inhibition of an enzyme.
In this equation, ki (which is analogous to kcat for a substrate) is the rate constant for the reaction that generates the inactivated form of the enzyme (EI*) once the enzyme is saturated by the irreversible inhibitor. A ki value is obtained by dividing the maximum rate of inactivation (V max for the inactivation reaction) by the total enzyme concentration. Ki is the concentration at which the inactivation reaction proceeds at 50% of the maximum velocity (analogous to Km for a substrate). The ratio of ki /Ki is the second-order rate constant for the reaction between free enzyme and free inhibitor to form the inactivated form of the enzyme. Note that the Ki value for an irreversible inhibitor is conceptually very different from the Ki value for a reversible inhibitor, meaning it is essential to clearly represent the class of inhibitor when reporting Ki values.
Ideally, irreversible inhibitors should have high ki values that indicate a rapid reaction between inhibitor and enzyme forming the covalent inhibitor–enzyme complex and low Ki values which generally reflect strong noncovalent preassociation of the inhibitor and the enzyme prior to the inactivation reaction. Inhibitors with large ki /Ki values bind to and react quickly with their target enzymes. Because the action of an irreversible inhibitor is time dependent, 31 measuring IC50 values is inappropriate since IC50 values are highly dependent on the substrate concentration and incubation time within an experiment. However, as mentioned above, there is a practical limit to irreversible inhibitor efficiency when used for radiotracer imaging. Irreversible inhibitors whose efficiency is above a certain limit (which is unique for each enzyme) produce images that actually reflect blood flow rather than enzyme activity. This situation has notably been observed for extremely rapid irreversible inhibitors of both acetylcholinesterase (AChE) 32,33 (discussed below) and MAO. 15 For irreversible inhibitors which are not so rapid as to be limited by blood flow, the ki /Ki value should reflect efficiency under physiological concentrations of enzyme and tracer levels of imaging agent. Often, radiotracer derivatives having varying ki /Ki values must be evaluated in vivo accompanied by kinetic modeling to account for blood flow to determine the ideal inactivation efficiency required for quantitative imaging experiments of a particular target.
Imaging Enzyme Activity in the Brain
Some hydrolytic enzymes that are of interest for imaging with PET/SPECT are located in the brain, meaning a small molecule tracer must access the central nervous system (CNS). All imaging agents, both in the CNS and in peripheral systems, generally need to be selective for their target, reasonably tight-binding to their target, moderately hydrophilic (to avoid widespread nonspecific binding, especially to lipids), reasonably resistant to metabolism, easily synthesized and radiolabeled, and safe at tracer levels (which is largely dependent on the specific activity of the injected tracer). Additionally, to image targets in the brain, a molecule must also be able to cross the BBB. To cross the BBB, a molecule generally must be relatively small (<500 g/mol and <80 Å cross-sectional area), moderately hydrophobic (log D 2.0 − 3.5), neutral, and have few hydrogen-bond donor and acceptor sites. Additionally, a probe must not be a substrate for efflux transporters such as P-glycoprotein (PgP) or breast cancer–resistance protein (Bcrp). 34 -36 Although progress continues to be made in the development of imaging agents for enzymes in the CNS, probe design remains an ongoing challenge. 37
EC 3.1: Esterases
Acetylcholinesterase and Butyrylcholinesterase
Acetylcholinesterase plays a critical role in nerve signaling, as it is responsible for the postsynaptic hydrolysis of the neurotransmitter acetylcholine following activation of the postsynaptic neuron. Molecular imaging of AChE is of interest to help clarify the role of the enzyme in Alzheimer disease progression, in particular. 34,38 Imaging of AChE has been well studied and an interested reader should consult one of the excellent reviews on recent progress in radionuclide-based molecular imaging for AChE. 38 -40 Since publication of those reviews, several more articles on PET imaging of AChE have appeared. 41 -47
More recently, butyrylcholinesterase (BChE) was identified as a potential imaging target since it also can play a role in acetylcholine metabolism, especially in patients with neurodegenerative diseases such as Alzheimer’s. 48 Specific imaging of BChE is a new area of interest, and recently agents specifically targeting BChE have been reported. 49,50
Lipase
Monoacylglycerol lipase (MAGL) is a serine hydrolase which catalyzes the postsynaptic hydrolysis of 2-arachidonoylglycerol (2-AG; see Figure 4), which is an endocannabinoid neurotransmitter. 52 2-Arachidonoylglycerol is synthesized by the postsynaptic neuron and diffuses to the presynaptic neuron where it activates cannabinoid receptors, leading to a reduction in presynaptic neurotransmitter release. The hydrolytic activity of MAGL is thought to be responsible for terminating approximately 85% of the signal from 2-AG. 53 Since problems with the endocannabinoid system have been suggested to play a role in a number of neurological diseases such as addiction, pain, anxiety, and schizophrenia, small molecule tools to modulate endocannabinoid activity without unwanted psychoactive effects are desirable both as therapeutics and probes of endocannabinoid function. 54 For these reasons, MAGL has recently been identified as a target of interest for imaging with PET/SPECT.
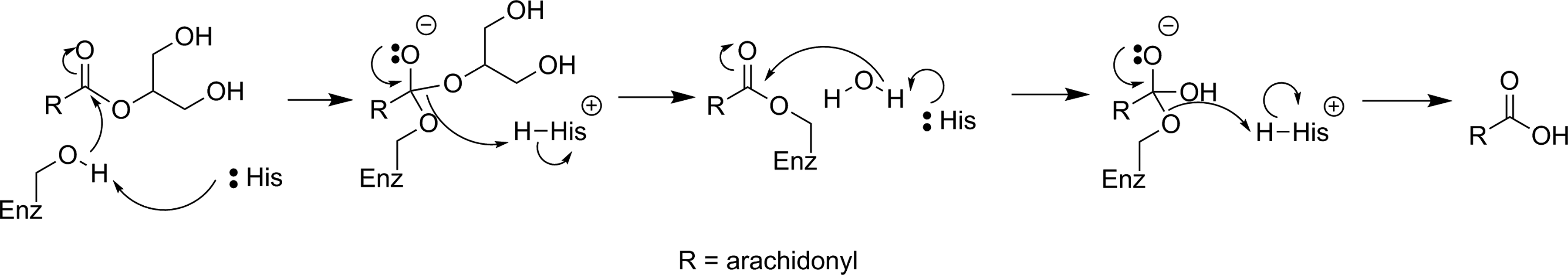
Mechanism for hydrolysis of 2-AG by MAGL. 51 2-AG indicates 2-arachidonoylglycerol; MAGL, monoacylglycerol lipase.
The first attempt to develop a PET imaging agent for MAGL was by Wilson and coworkers, who prepared a total of 12 putative irreversible inhibitors for MAGL (7 known compounds and 5 novel molecules).
55
These irreversible inhibitors were designed to tag the catalytic nucleophilic serine of MAGL through reaction with an activated carbamate or urea moiety present in the inhibitor to form a very stable covalent–enzyme intermediate (see Figure 5). To estimate inhibitor potency and specificity, IC50 values were measured for all 12 compounds as inhibitors of both MAGL and fatty acid amide hydrolase (FAAH), which is a related enzyme responsible for terminating endocannabinoid signaling through hydrolysis of anandamide, as discussed below. A further complicating factor in development of an imaging agent for MAGL in the brain is the modest level of MAGL activity in the blood that could potentially lead to high background signal by nontarget tracer binding. Five of the inhibitors (

Reaction of MAGL with carbamate-based (X = O) or urea-based (X = NH) irreversible inhibitors to form a stable enzyme–inhibitor complex. MAGL indicates monoacylglycerol lipase.
Carbamates (

Reaction of [11C]CO2 to form carbamates (X = O) or ureas (X = NR). *11C. 56
Recently, 2 independent radiosyntheses were reported for [11C]SAR127303 (
Recently reported 11C-labeled irreversible inhibitors for MAGL. *11C. MAGL indicates monoacylglycerol lipase.
Phosphodiesterases
Cyclic nucleotide phosphodiesterase (PDE) enzymes are a family of 11 enzymes that play an important role in termination of cellular signaling by hydrolyzing the secondary messenger molecules cyclic adenosine monophosphate and cyclic guanosine monophosphate. Some members of this family of enzymes (PDEs 2, 4, 5, 7, and 10) have been identified as potential targets for PET imaging, especially in the brain. There has been a substantial amount of work in the preparation and testing of potential PET/SPECT imaging agents for PDEs, which have recently been covered in 2 excellent and comprehensive reviews covering developments prior to 2012 60 and between 2012 and 2016. 61
Sulfatases
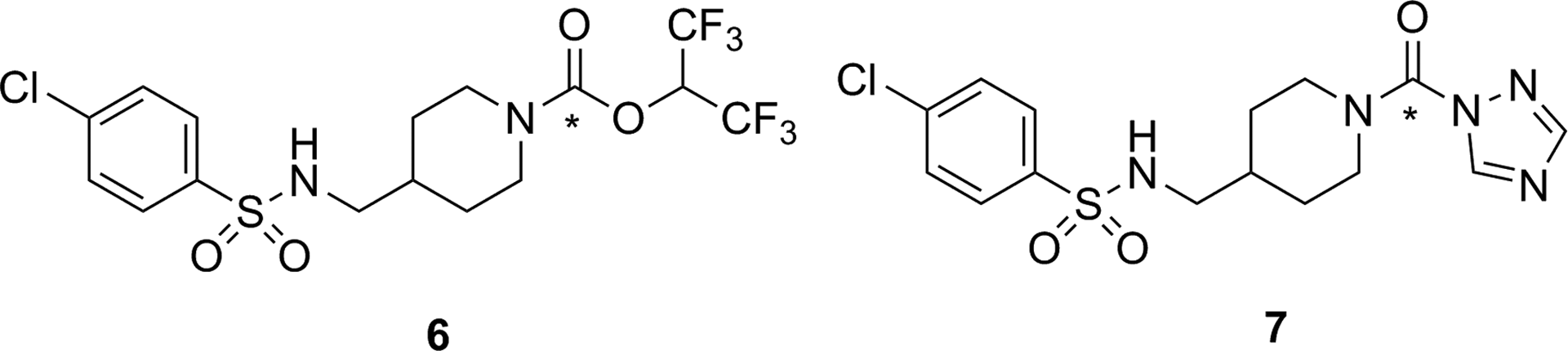
Steroid sulfatase (STS; estrone sulfatase) has been identified as a potential target for imaging with PET/SPECT. Increased STS activity has been demonstrated in some hormone-dependent breast cancers: The enzyme cleaves sulfate from biologically inactive estrogen-3-sulfates (estrone-3-sulfate, estradiol-3-sulfate, or estriol-3-sulfate) to release the desulfonated estrogen (see Figure 9), which in turn increases tumor growth by activating the estrogen receptor. The aim of developing an imaging agent for STS is to visualize and differentiate breast cancer subtypes. Given that other targets such as estrogen receptors already allow PET/SPECT imaging of breast cancer subtypes, 63 there is not a pressing need for development of an imaging agent targeting STS.
Proposed mechanism for STS hydrolysis of an estrogen-3-sulfate. 62 STS indicates steroid sulfatase.
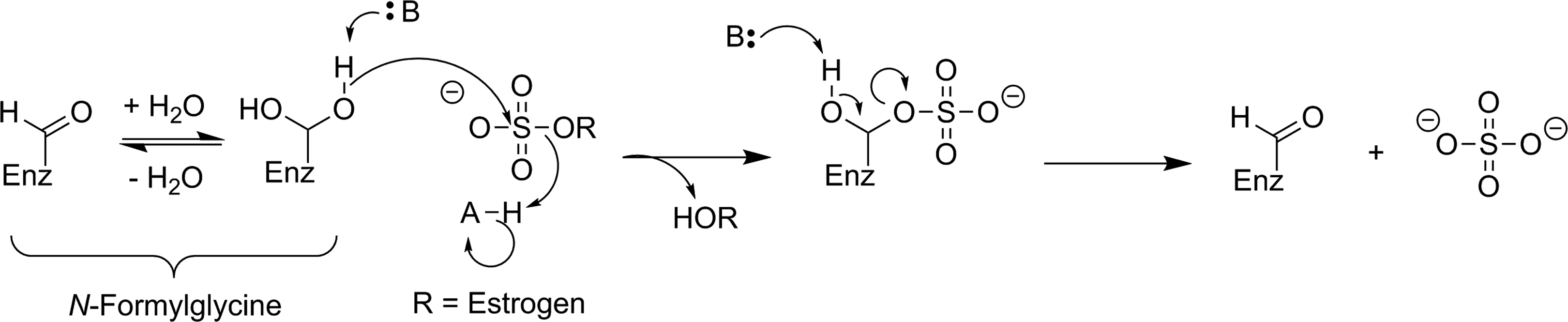
There have been a few attempts to image STS activity, though no successes have been reported. All of the reported radiolabeled probes for STS are irreversible inhibitors with an aryl sulfamate moiety. While the mechanism for irreversible inhibition of STS with an aryl sulfamate has not been definitively proven, these inhibitors have been proposed to function through formation of an imine N-sulfate with the catalytic formylglycine residue (Figure 10).
62,64,65
However, carbonic anhydrase (CA, EC 4.2.1.1) is also sensitive to inhibition with aryl sulfamates, meaning that selectivity between these 2 enzymes is an important consideration for development of a nuclear imaging agent.
65
Derivative [18F]

Proposed mechanism of irreversible inhibition of STS by a sulfonamide through imine N-sulfate formation with the active-site formylglycine residue. 62 STS indicates steroid sulfatase.
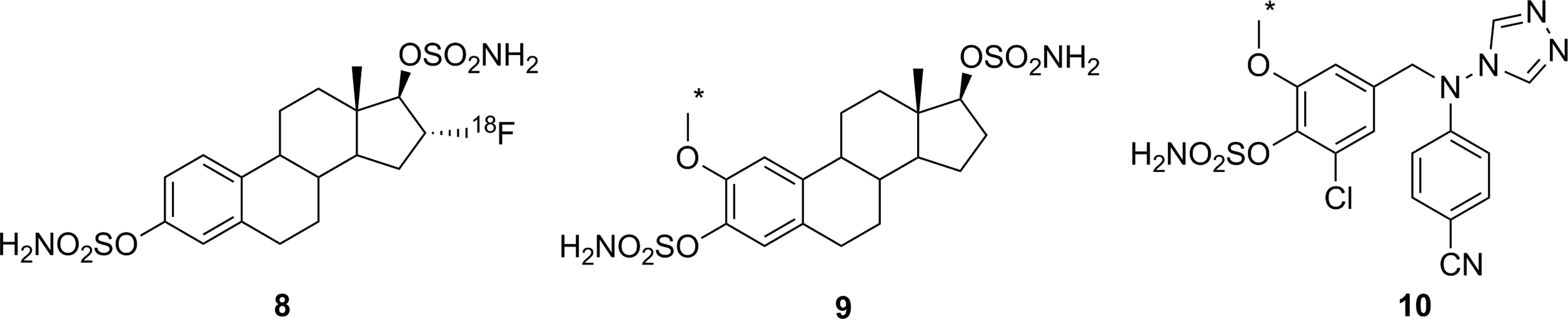
Irreversible inhibitors prepared as potential PET imaging agents for STS. *11C. PET indicates positron emission tomography; STS, steroid sulfatase.
EC 3.2: Glycosylases
Glycosidases
Glycoside hydrolases are a large family of enzymes responsible for hydrolytic cleavage of 1 or more sugar residues from a variety of biomolecules, including oligosaccharides or polysaccharides, peptides or proteins, and lipids. Glycosidases have been shown to play important roles in a number of diseases, including diabetes, 70 Parkinson disease, 71 cancer, 72 and metabolic disorders such as Gaucher disease. 73 However, despite their biological importance, only moderate progress has been made in developing PET/SPECT imaging agents for glycosidases, despite a number of attempts using different approaches.
Three retaining β-glycosidases have frequently been targeted as potential nuclear imaging markers: Escherichia coli β-galactosidase (lacZ), human lysosomal β-glucuronidase (GUS), and human β-glucocerebrosidase (acid β-glucosidase, GBA1). Figure 12 shows a generic retaining β-glycosidase mechanism. LacZ is an enzyme of interest owing to its frequent use as a reporter gene; an imaging agent could be used to identify expression of a gene of interest in a whole organism in a noninvasive way. GUS, normally restricted to the lysosome, is overexpressed in the extracellular tumor microenvironment and is a potential imaging target for tumor localization. GBA1 is also a lysosomal enzyme whose deficiency leads to Gaucher disease. 75 More recently, mutations in the gene encoding for GBA1 have also been identified as the single largest genetic risk factor for the development of Parkinson disease, and low levels of GBA1 enzyme activity found in sporadic, early, and late Parkinson patients make it a potential diagnostic and/or therapeutic target. 76

General retaining β-glycosidase mechanism. 74
Four compounds (

Compounds tested as substrate-based nuclear imaging agents for lacZ (

Proposed mechanism for radiotracer accumulation following hydrolysis of
Efforts to image GUS activity using metabolic trapping following hydrolysis of a substrate have been more successful. Since GUS activity is found in the extracellular tumor microenvironment,
81
efficient membrane permeability is not a concern during probe design. Indeed, having a membrane impermeable tracer would avoid ubiquitous low-level GUS activity in the lysosome and high normal tissue uptake, so only tumor tissues with high extracellular GUS activity will be detected. However, radiotracer substrates intended for imaging of GUS activity also have the drawback that glucuronylation is a common in vivo modification of xenobiotics to increase solubility prior to renal excretion, meaning that glucuronylated tracers are highly likely to accumulate in the kidneys and bladder. Compound
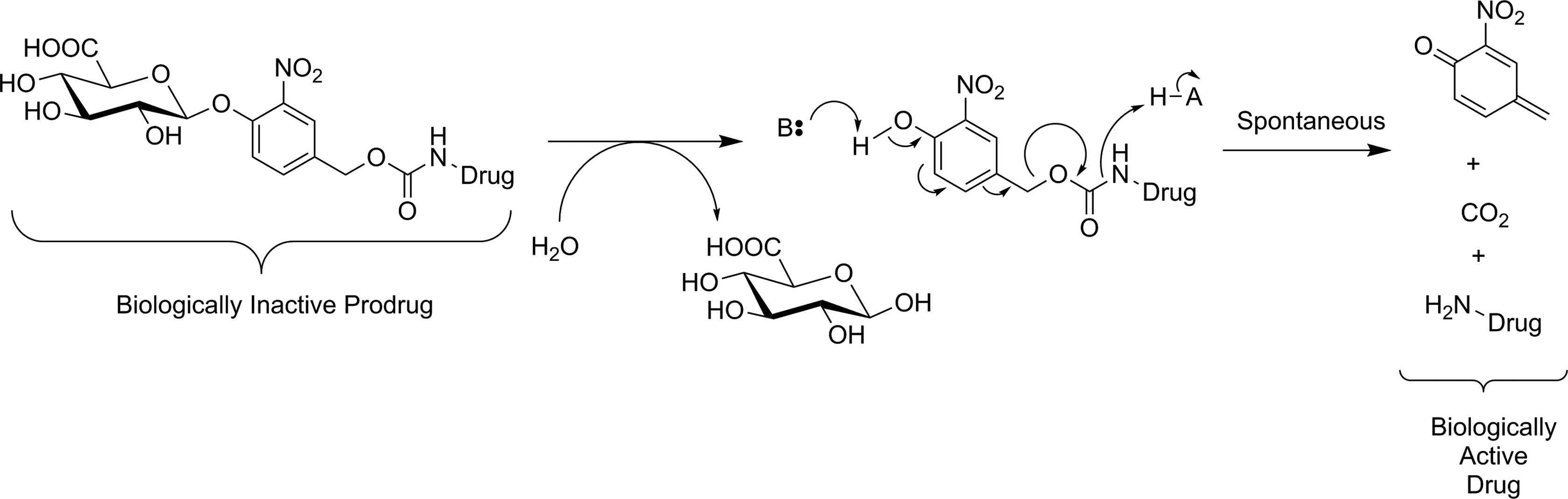
Activation of a glucuronide prodrug conjugate by GUS. The active form of the drug is released through spontaneous decomposition of the self-immolative linker following enzyme-mediated hydrolysis. 84 GUS indicates β-glucuronidase.
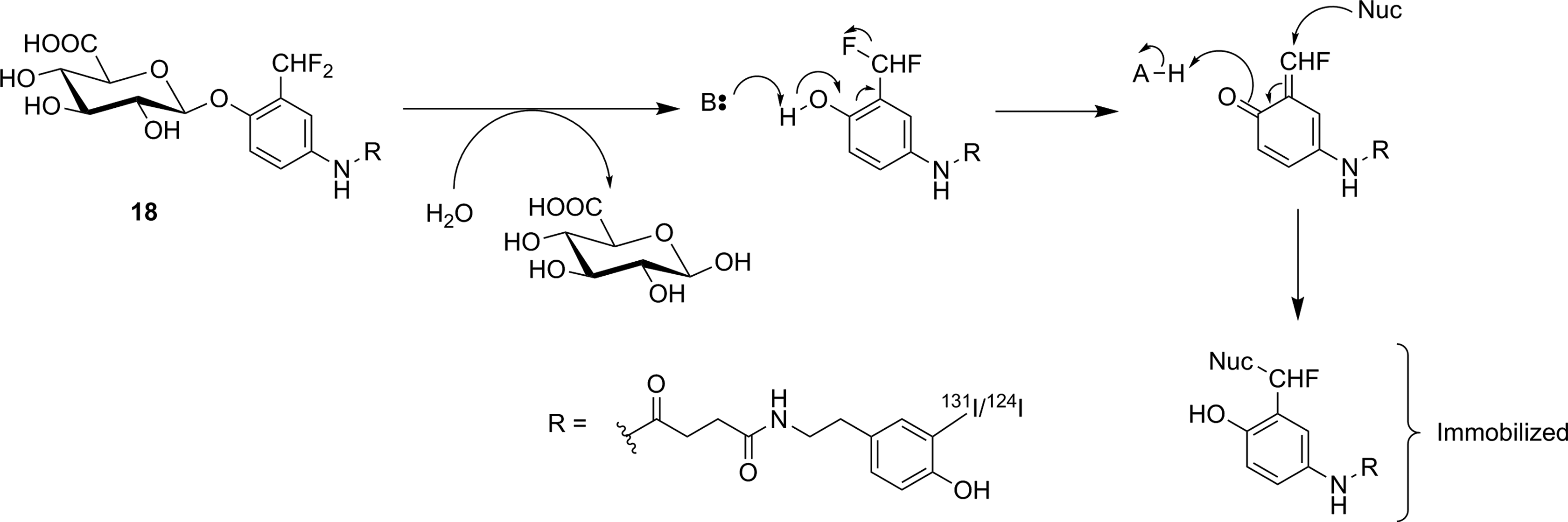
Hydrolysis of
Radiolabeled reversible glycosidase inhibitors have also been prepared as potential PET/SPECT imaging probes. Nojirimycin (
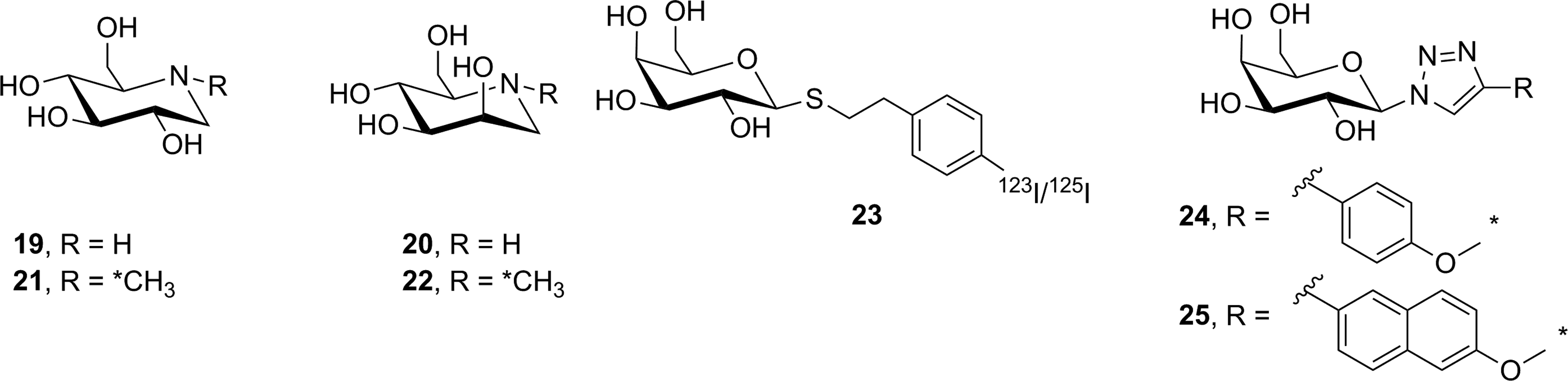
Reversible inhibitors prepared as potential glycosidase imaging agents. *11C.
To date, 3 reversible inhibitors targeting lacZ (
The Withers group has shown that fluorosugars with good leaving groups (either fluoride or dinitrophenol) at their anomeric center are effective irreversible inhibitors for many retaining glycosidases.
94
They inhibit enzyme activity by forming a long-lived fluorosugar–enzyme covalent conjugate (Figure 18). Fluorosugar inhibitors for retaining β-glucosidases typically have a 2-deoxy-2-fluoro modification, making them structurally similar to FDG. However, in only 1 instance (discussed below), has [18F]FDG served as the starting material for the synthesis of a fluorosugar inhibitor. [18F]2-Fluoro-2-deoxyglucose has not been the general starting material for fluorosugar inhibitors because the nonradioactive versions of such inhibitors are prepared through a nucleophilic substitution reaction at the anomeric center with a highly activated leaving group (to prepare the glycosyl fluoride) or a nucleophilic aromatic substitution reaction with the free sugar hemiacetal (to prepare the dinitrophenyl derivative). The time needed to selectively protect the other hydroxyl functional groups, perform the desired reaction at the anomeric center, and deprotect the other hydroxyl groups is incompatible with the rapid reactions required for short-lived isotope radiosynthesis. Therefore, early efforts to prepare fluorosugar glycosidase irreversible inhibitors ([18F]

Irreversible inhibition of a retaining β-glycosidase by an activated 2-deoxy-2-fluoro glycoside. X = fluorine or O-dinitrophenyl. 94

Irreversible inhibitors investigated as GBA1 imaging agents. Note that an individual molecule of [18F]

Reaction between glucal and [18F]F2 to form [18F]
In an attempt to address some of these issues, a β-gluco-configured 6-[18F]fluoro analogue ([18F]
EC 3.3: Ether Hydrolases
Epoxide Hydrolase
Recently, the development of a potential imaging agent for soluble epoxide hydrolase (sEH) has been reported.
99
This enzyme catalyzes the degradation of epoxyeicosatrienoic acids, which are important signaling molecules for controlling blood flow in the brain. Inhibitors for this enzyme such as

Known inhibitor for soluble epoxide hydrolase (
EC 3.4: Proteases
Given their wide variety of critical biological roles, proteases are understandably a family of enzymes that have received a great deal of attention in the development of PET/SPECT imaging agents. All of the proteases of interest that have been investigated as potential nuclear imaging targets have been well studied and also reviewed recently. As a consequence, the treatment of those topics here will be cursory and will instead direct the interested reader to appropriate review articles or reports recently published.
Cathepsins
Cathepsins are proteases that are often (though not universally) lysosomal enzymes involved in catabolic degradation of cellular components. Their dysfunction has been linked to a large number of diseases, including cancer, Alzheimer disease, arthritis, stroke, and parasitic infections. 102 The PET/SPECT probes for cathepsins have been recently reviewed, 103 and only a few articles on nuclear imaging of cathepsins have appeared since. 104 -106
Prostate-Specific Membrane Antigen (Glutamate Carboxypeptidase 2)
Prostate-specific membrane antigen (PSMA; glutamate carboxypeptidase 2) is a membrane-bound metalloprotease that is highly upregulated in cancerous prostate cells, making it a very attractive target for imaging of primary and metastatic tumors. A large number of PET/SPECT imaging agents for PSMA have been described, and many excellent reviews have been published. 107 -109 Currently, prospective imaging agents for PSMA have advanced to small-scale clinical studies, hopefully leading to large-scale studies to validate their use in the diagnosis of metastatic prostate cancer. 110
Caspases
Caspases (cysteine-dependent aspartic proteases) are a family of cysteine proteases that play a key role in apoptosis, or programmed cell death, and in inflammation. Although lacking specific discussion of PET/SPECT imaging agents, a well-written comprehensive review on general probes for caspases, especially focusing on optical imaging agents and inhibitor development, has recently been published. 111 However, a recent review from 2015 has surveyed PET/SPECT imaging agents for caspases 3 and 7. 112 Since publication of that review, there have been several articles published on nuclear imaging agents targeting caspases. 113 -119
Matrix Metalloproteases
Matrix metalloproteases (MMPs) are a large family of Zn2+-dependent endopeptidases primarily responsible for degradation of a variety of proteins in the extracellular matrix. They are involved in a wide variety of important biological processes, including apoptosis, cell proliferation and migration, differentiation, and angiogenesis. Owing to their upregulation in many cancers, MMPs 2 and 9 (gelatinases) have been the enzymes of most interest. A summary of advances in the development of potential imaging agents for MMPs was last published in 2013, 120 and since then, there have been other reports of inhibitors 121 -132 and substrates 133,134 designed to image MMP expression and activity, respectively. However, despite the considerable efforts to design an imaging agent for MMPs, to date, no imaging agent has made it into human trials.
EC 3.5: Hydrolases Acting on Nonpeptidic Carbon–Nitrogen Bonds
Histone Deacetylase
There are 4 classes of histone deacetylase (HDAC) enzyme: classes 1, 2, and 4 are zinc-dependent enzymes, while class 3 enzymes (also referred to as sirtuins) are NAD+ dependent. 135 These enzymes are responsible for cleaving an acetyl moiety from the ∊-nitrogen on the side chain of a lysine in a histone protein. Figure 22 depicts a general reaction mechanism for a Zn2+-dependent (class 1, 2, or 4) HDAC. Protonation of the resulting free amine causes negatively charged DNA to wrap around the histone protein, thereby silencing expression of nearby genes, meaning HDACs are one mechanism for epigenetic control of gene expression. 135 They have also been shown to act on a number of nonhistone proteins, meaning they have broader functions than just control of DNA expression. 137
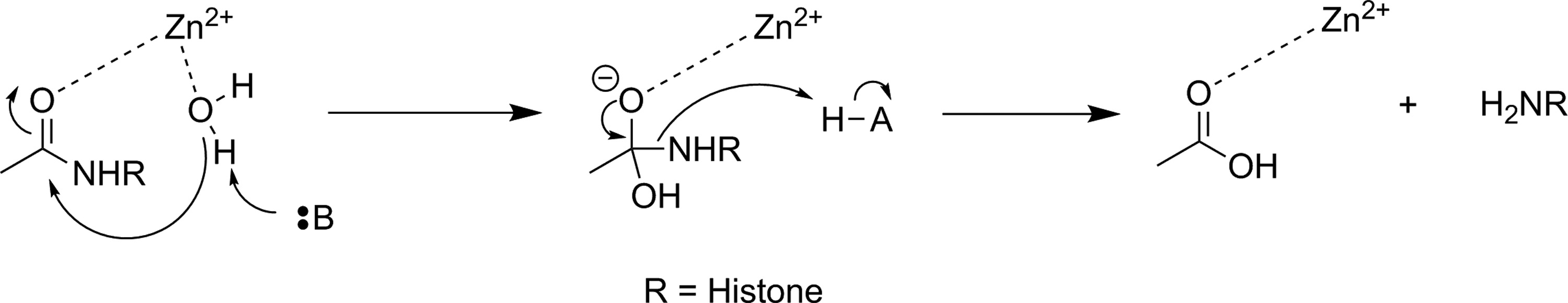
Simplified mechanism for a Zn2+-dependent HDAC. 136 HDAC indicates histone deacetylase.
Altered HDAC levels have been detected in neurological degenerative diseases, such as Alzheimer disease, bipolar disorder, schizophrenia, major depressive disorder, stroke, epilepsy, and multiple sclerosis. 137 Because of their role in epigenetic regulation, HDACs have also been linked to cancer. 135 Class 1 and 2 HDAC activities have been particularly linked to disease and have been best studied. Class 1 enzymes are primarily found in the nucleus, while class 2 are much larger and can move between the nucleus and the cytoplasm. Class 2 can be further subdivided into 2a and 2b, which are both important for a variety of disorders, most notably various cancers. 135 A tracer able to image variations in HDAC levels in both healthy and diseased individuals will be useful for better understanding the role of these enzymes in epigenetics, as well as studying the efficacy of potential therapeutics. It is important that such an imaging agent is able to cross the BBB, since many of the diseases of interest are brain disorders.
Efforts to develop imaging agents for HDACs have focused on producing substrates and noncovalent inhibitors; no irreversible inhibitors have been described to date. The first attempt to image HDAC activity used 6-([18F]fluoroacetmido)-1-hexanoicanilide ([18F]FAHA or [18F]

Potential substrate-based imaging agents for HDACs. *11C. HDACs indicates histone deacetylases.
Approaches employing reversible inhibitors of HDAC have shown varied success. All of the potential imaging agents based on reversible inhibition of HDAC have employed a functional group (hydroxamate or carboxylic acid) designed to coordinate to the active-site zinc ion found in class 1 and 2 HDACs. Known HDAC inhibitors have served as the starting scaffold for most radiolabeled HDAC inhibitors reported to date. For example, 11C-labeling of the pharmaceuticals butyric acid ([11C]
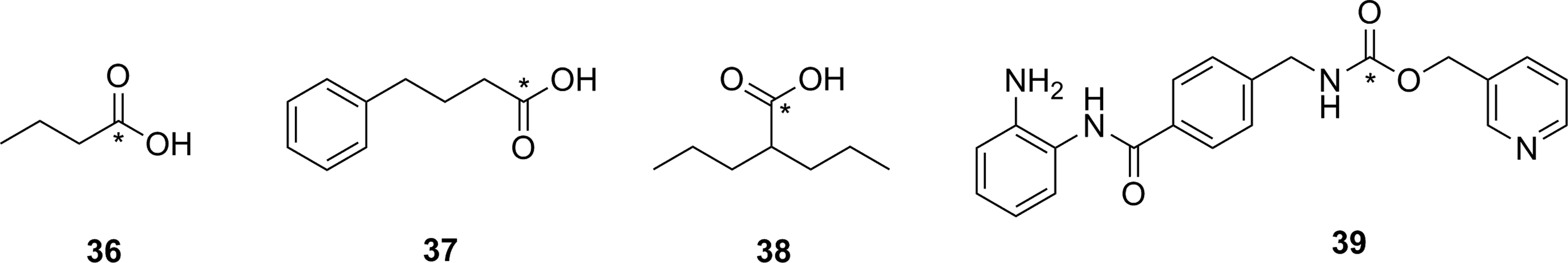
Potential imaging agents for HDAC based on clinical or preclinical drugs. *11C. HDAC indicates histone deacetylase.
Derivatives of another compound in clinical use, suberanilohydroxamic acid (SAHA, vorinostat,

Suberanilohydroxamic acid (
Hooker and coworkers have made the greatest progress toward an imaging agent of HDAC expression suitable for clinical use with Martinostat ([11C]

Martinostat (
There have been three other radiolabeled inhibitors of HDAC that have been investigated as potential imaging agents. Compound [11C]

Other HDAC inhibitors and potential imaging agents. *11C. HDAC indicates histone deacetylase.
Fatty Acid Amide Hydrolase
Along with MAGL (discussed above), FAAH is the other hydrolase enzyme known to play a major role in the endocannabinoid neurotransmitter system. The most important natural substrate for FAAH is thought to be anandamide (N-arachidonoylethanolamine), which is a retrograde lipid neurotransmitter 51 and whose hydrolysis is depicted in Figure 28. Note that although FAAH is a serine hydrolase, it uses an unusual Ser-Ser-Lys catalytic triad. 159 Inhibitors of FAAH are being investigated as tools to better understand the role of FAAH in disease or as potential therapeutics for conditions such as addiction, anxiety, schizophrenia, depression, and neurodegeneration. 160,161 From a nuclear imaging perspective, FAAH is an interesting target since it is an intracellular target with a very hydrophobic substrate requiring a specific transport protein. 162 Small molecule probes attempting to image FAAH activity and distribution have been developed using all 3 types of probe: substrates, reversible inhibitors, and irreversible inhibitors.
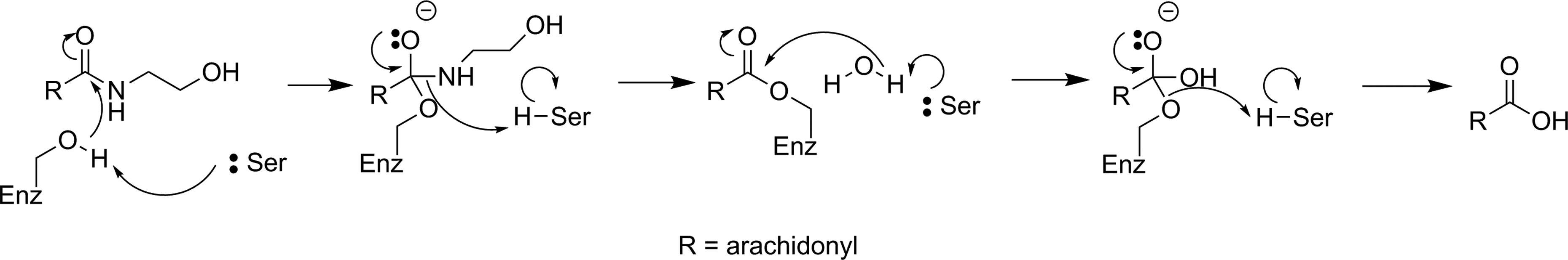
Mechanism for hydrolysis of anandamide by FAAH. 159 FAAH indicates fatty acid amide hydrolase.
The first attempt to image FAAH used 123I-labeled arachidonic and linoleic acid amide analogues (

Potential substrates for SPECT (
Most of the irreversible inhibitors for PET imaging of FAAH contain a carbamate or urea group that acylates the nucleophilic serine residue in the active site (Figure 30).
166
The first attempt to image FAAH using an irreversible inhibitor was inspired by the known carbamate-containing irreversible inhibitor URB597 (

Reaction of FAAH with carbamate-based (X = O) or urea-based (X = NH) irreversible inhibitors to form a stable enzyme–inhibitor complex. 166 FAAH indicates fatty acid amide hydrolase.

Known carbamate irreversible inhibitor for FAAH (
In addition to [11C]

Other carbamate-based irreversible inhibitors for FAAH as potential PET imaging agents for FAAH. *11C. FAAH indicates fatty acid amide hydrolase; PET, positron emission tomography.
Four urea-based irreversible inhibitors of FAAH have been reported as potential imaging agents for FAAH. [11C]
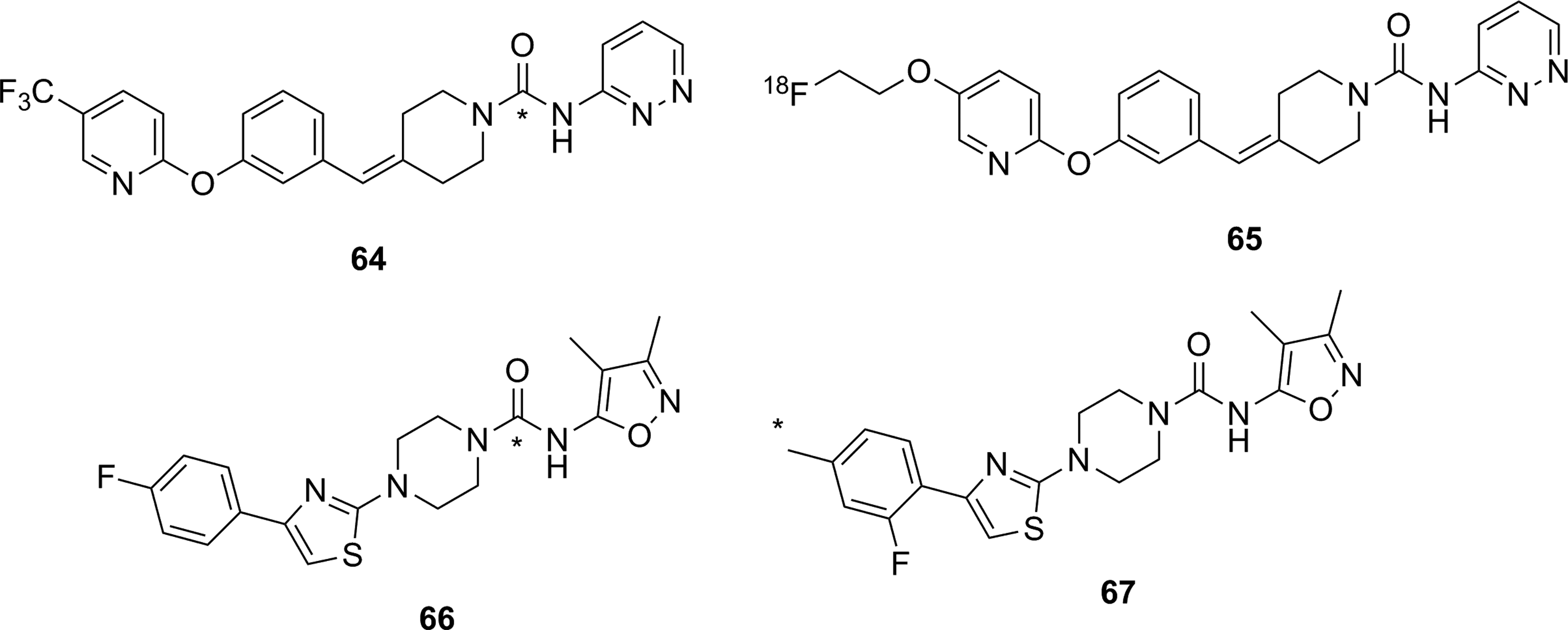
Urea-based irreversible inhibitors for FAAH as potential PET imaging agents for FAAH. * 11C. FAAH indicates fatty acid amide hydrolase; PET, positron emission tomography.
To date, three radiolabeled reversible inhibitors of FAAH have been reported as imaging agents of FAAH. The first two compounds, [11C]
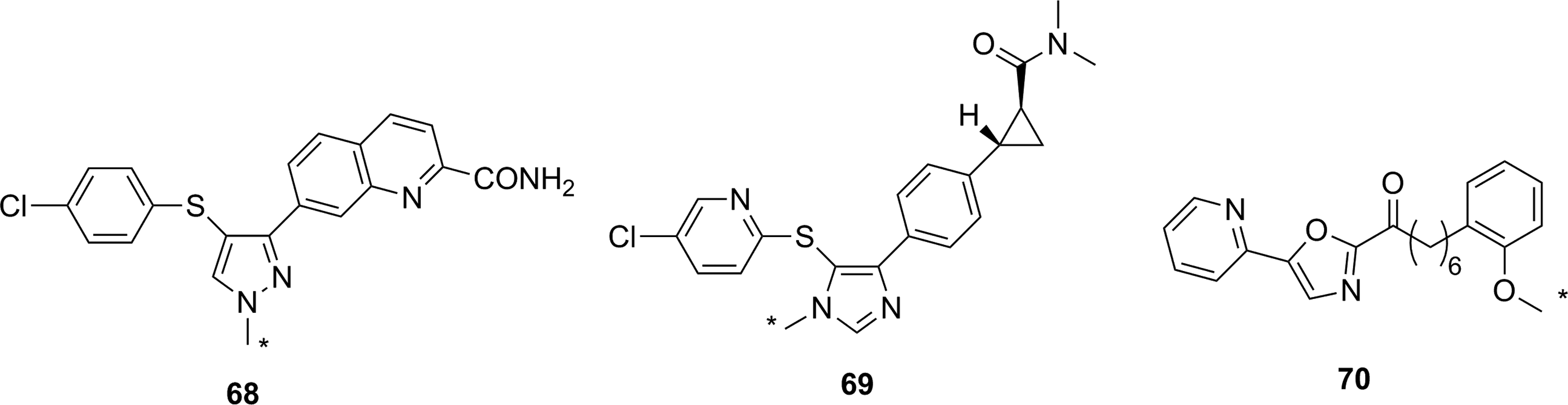
Noncovalent inhibitors used as PET imaging agents for FAAH. *11C. FAAH indicates fatty acid amide hydrolase; PET, positron emission tomography.
Conclusions and Perspectives
As described in this review, the most successful imaging agents have followed a development process somewhat akin to the following: Identify an enzyme target linked to an important biological process (such as a disease) as a valuable imaging target. Select a lead compound as a potential tracer. This is either through rational design of a new substrate/inhibitor molecule (such as Martinostat, [11C] Measure the enzyme kinetics for the lead compound as a substrate, reversible inhibitor, or irreversible inhibitor in vitro. Specificity for the target enzyme is verified by demonstrating that the probe candidate is not efficiently recognized by similar or related enzymes through an in vitro screen using recombinant enzymes, cell lysate, or cell assays. Optimize the lead compound structure for desirable properties such as in vitro efficiency and selectivity. Perform cell-based studies for membrane permeability and intracellular target efficiency and selectivity. Such experiments can be greatly aided by employing genetic knockout techniques to create models that are either positive or negative for a given enzyme or small molecule inhibitors to reduce target activity in cellulo. For enzyme targets in the brain, performing an assay to model BBB permeability is also desirable.
191,192
Prepare a radiolabeled analogue of the tracer candidate using convenient radiolabeling chemistry with sufficient radiochemical yields to provide enough tracer in high purity and specific activity for small animal studies. Perform ex vivo biodistribution studies in a small animal model (typically a rodent) to determine modes and rates of clearance for the tracer, metabolic stability, and organ distribution and tissue or tumor uptake (in the case of oncology-based tracers). Measure the specificity of the radiotracer for the target enzyme in vivo through co-injection of a nonradioactive analogue of the tracer with known binding to the target to saturate the target enzyme and block processing and/or retention of the tracer. Additional experiments to confirm specificity in vivo are helpful, such as correlating tracer uptake with direct measurements of enzyme expression levels (eg, Western blots) as well as using well-established co-injection of a known selective inhibitor of the target enzyme. Ex vivo tissue samples can also be evaluated by overlaying images from autoradiography and immunohistochemical staining to confirm co-localization of the target enzyme with the radioactive tracer. The PET/SPECT imaging in a small animal model, both a disease model and a healthy control. Imaging results should be correlated with biodistribution studies, and additional blocking studies can also be performed with PET/SPECT imaging. Especially for target enzymes found in the brain, metabolism and imaging studies with nonhuman primates. Begin first in human studies, potentially advancing to clinical human trials.
2
These development steps represent an ideal process that may not be feasible in all cases. Of course, careful planning of future steps can influence decision-making earlier in the process. For example, consideration of a planned radiosynthetic route can help guide the earlier, structure–activity optimization process. One common theme in the development of tracers that ultimately proved unsuccessful was a lack of experiments done in vitro and in cellulo to identify potential problems. Although tracer candidates that have shown great promise by thorough characterization in vitro are not guaranteed to have great in vivo imaging success, careful screening of early candidates can rule out poor candidates prior to expensive and often time-consuming radiolabeling and unnecessary animal studies (which has ethical considerations). In many of the unsuccessful attempts at radiotracer development for hydrolytic enzymes that failed in vivo, subsequent in vitro experiments revealed weaknesses and deficiencies that could have been identified prior to animal studies. For example, complete and appropriate enzyme kinetic evaluation of a tracer candidate should be performed prior to in vivo study to identifying unsuitable candidates, as well as in cellulo experiments to verify membrane permeability for intracellular targets or BBB permeability for targets in the brain. Additionally, in vitro experiments using purified enzymes to carefully delineate tracer specificity among isozymes would likely increase chances for success in vivo.
The majority of nuclear imaging agents for hydrolytic enzymes reported to date are reversible or irreversible inhibitors. This is particularly true for proteases (not discussed in detail herein). In contrast, there have been many fewer substrate-based imaging agents reported, and even fewer of those have proven successful. Unlike the development of an inhibitor, for which an in vitro enzyme assay is already established, most new substrates do not have kcat and Km values, nor experiments testing specificity over related enzymes, reported in the literature. Indeed, there are numerous difficulties and challenges in developing a new assay, such as using substrates without chromophores or fluorophores, solubility issues of some enzymes (ie, membrane bound or associated enzymes), and a lack of availability of purified enzymes. This is an unfortunate gap in knowledge: If a putative substrate-based imaging agent has suitable pharmacokinetic properties yet is unable to image the target enzyme, it can be difficult to determine whether the failure is a result of the substrate being kinetically incompetent at tracer levels or is processed by a similar (but off-target) enzyme. Another major challenge for substrate-based imaging agents has been the lack of a reliable metabolic or chemical immobilizing moiety that traps the radionuclide at the site of enzyme activity. Strategies relying on either hydrophobicity to lock the radioactive reporter in cell membranes or precipitation or the unmasking of an ionizable group to generate a cell membrane impermeable ion are clever, but this makes development of new tracers even more complicated and these methods have been met with variable success. Nonetheless, the development of a reporter group that can be readily attached to a substrate, does not interfere with recognition by the target enzyme, is easily radiolabeled, and reliably accumulates at the site of enzyme action would be a major advancement in developing substrate-based nuclear imaging agents for hydrolytic enzymes.
Just as the development of [11C]CO2 fixation technologies 56,193,194 led to a surge in development of imaging agents for both HDAC and FAAH, other new radiosynthetic technologies are likely to drive the development of new tracers for imaging a wide array of hydrolytic enzymes. Exciting and convenient (“kit-like”) new approaches have been recently reported that could easily drive the future development of tracers for imaging hydrolytic enzymes, such as reactions with uncommon nuclei 195 including [18F]trifluoroborates, 196 -198 [18F]silicon fluoride, 199,200 [18F]sulfonyl fluoride-containing prosthetic groups, 201 and chelation of Al[18F]F. 202 The major drawback to many of these techniques as they currently exist is that they require attachment of prosthetic groups or chelators to facilitate radiolabeling, which are chemical moieties that add steric bulk and can change the polarity and biodistribution of the attached drug. Another promising area of future development are new reactions for rapidly forming aryl carbon–[18F]fluorine bonds. 203 -205
This is an exciting time in PET/SPECT radiotracer development for hydrolytic enzymes. There are now enough successful examples of tracer development to allow some reflection on promising strategies and an ideal process can be described. The development of new radiochemical methods will continue to drive innovation in tracer development. Finally, the rapid and successful development of imaging agents for both HDAC and FAAH shows the value of careful and thorough experimentation at each step of the development process. As well, involvement of a multidisciplinary development team that includes synthetic organic chemists, enzymologists, radiochemists, biologists, imaging scientists, and clinicians now appears to be critical for rapid success in taking an imaging agent from concept to human trials.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.