
Abstract
Like Janus, the Roman god of beginnings, transitions, and endings, spreading depolarizations (SDs) can be depicted with two faces: one looking backward, waving a symbolic farewell to the end of a cortical seizure; the other forward looking, opening a darker door for a fatal wave in the brainstem that ends life. There is good agreement on the distinct electrical nature of both events, but neither role is yet proven in patients. SD is a slow-moving wave of cellular depolarization that steadily silences neuronal networks and depresses EEG amplitude, whereas seizures represent fast, intermittent synchronization of neural networks with highly variable EEG activation patterns. However, the thresholds triggering both events are neither fixed nor inseparable; indeed, their co-occurrence and interaction depend on dimly-lit intrinsic brain pathophysiology. New insights into single gene control of SD and seizure thresholds are beginning to illuminate the darkness. Here, we review recent data and consider the title's question at the end.
Keywords
Introduction
By every definition, episodes of spreading depolarization (SD) and seizures are distinct brain excitability events with independent, yet genetically overlapping, dynamic thresholds. SD is a self-regenerative wave of cellular depolarization of both neurons and glia involving severe loss of transmembrane ionic gradients (eg, Na+, K+), which inactivates many ion channels and exhausts the energy substrate, resulting in neuronal silence for minutes. The SD wave, initially discovered as “spreading depression” of cortical activity by Leao in rabbit cortex 1 and later proposed as the basis of migraine aura, is firmly associated with vascular pathology and brain injury. It's clinical symptom in the occipital cortex, a traveling 3 mm/min “scintillating” scotoma beautifully self-mapped by Karl Lashley in 1941, 2 reflects transient network hyperexcitability at the wave's leading edge followed by minutes-long neuronal silence. Functional MRI imaging precisely correlates the spreading wavefront with serial depression of visual evoked responses. 3 While the slow speed of SD is reminiscent of Hughlings Jackson's focal “march” across the motor homunculus, and its postictal reversibility is similar to the neurological depression of Todd's paralysis, SD has long been overlooked as an inherent feature of epileptic brain. This omission is due to our inability to detect a slow cortical DC shift in clinical scalp EEG studies that rely on AC-coupled high-pass filtered signals. In such recordings, an SD wave may be accompanied by a visible decrease in EEG amplitude and frequency band; 4 however, this signature alone, without a spread to adjacent electrodes, is insufficient mechanistic proof. Valid electrophysiological evidence of SD in human brain requires unfiltered intracranial electrode recordings, as now typically employed during ICU post-stroke and brain injury monitoring where SD arises subsequent to hypoxia/ischemia or hemorrhage. 5 Despite this impediment, the appearance of SD in uninjured hyperexcitable epileptic brain and its potential impact on neurological function is beginning to be recognized.
Monogenic Overlap of SD and Epilepsy
A shared molecular pathogenesis of SD and epilepsy was not appreciated until the first evidence of genetic overlap was revealed by familial hemiplegic migraine type 1 (FHM1), an inherited syndrome due to mutation of CACNA1A, the pore-forming subunit of PQ calcium ion channels. 6 Functional analysis of Cacna1a variants in mouse models revealed that PQ channel loss-of-function variants raise SD threshold, 7 while gain-of-function mutations lower it. 8 Interestingly, both precipitate seizure disorders of different types; gain of PQ function enhances glutamate release leading to convulsive seizures, SD, and premature mortality, while the loss of function leads to absence seizures, no SD, and suppression of convulsive seizures in Kv1.1 KO mice. 9 A different pattern is seen in SCN1A (FHM3) sodium channel mutant mice, where both loss and gain of function mutations are associated with convulsive seizures and SD events.10,11 Other FHM genes, ATP1A2 (FHM2)12,13 and PRRT2,14,15 as well as a non-FHM gene Cadasil/Notch3, 16 are linked to developmental epilepsy phenotypes with variable paroxysmal motor features and SD patterns.
Distinctive Gene-Specific Patterns of Seizure and SD Thresholds
Long-term DC monitoring of monogenic developmental epileptic encephalopathy mouse models has identified additional genes that share a dual phenotype of lowered seizure and SD thresholds. In Kcna1 KO and Scn1a LOF mice with generalized seizures, the cortical SD waves are unilateral and may be tightly or only loosely coupled to a seizure event and may even outnumber seizures (Figure 1A&B).10,17 In stark contrast, mice with conditional Kcnq2 KO and Scn8aN1768D/+ GOF mutant mice (with enhanced persistent sodium current) both predominantly generate tightly coupled bilateral seizure-SD complexes where SD initiates even before seizure termination17,18 (Figure 1C&D). These bilateral depolarizations are pharmacologically facilitated by a KCNQ/M-current inhibitor and suppressed by a persistent Na+ current inhibitor, respectively. These models suggest that an imbalance of persistent cationic currents (ie, INaP and IKM) contribute to this “short seizure-SD interval” SD phenotype, indicating that the seizure-SD interaction is not stochastic and can be targeted pharmacologically.
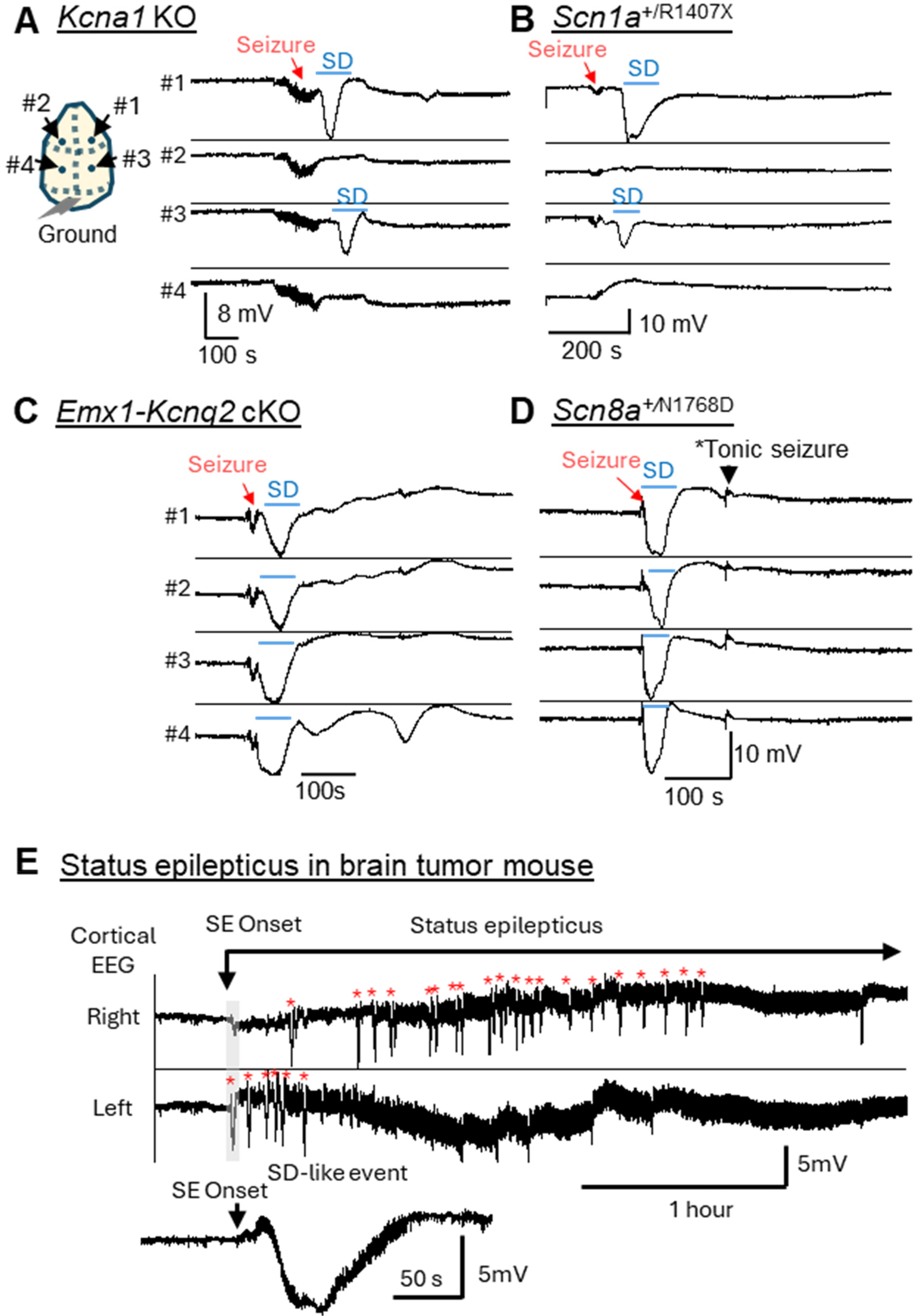
Generalized seizure with unilateral SD in Kcna1 KO and Scn1a+/R1407X mouse cortex. (A) In Kcna1 KO mice, postictal SD appears after seizure termination in anterior electrode, spreading to posterior electrode. (B) In Scn1a+/R1407X mice, a unilateral SD appeared 1 min after seizure termination first detected in posterior and subsequently in the anterior electrode. (C, D) Bihemispheric seizure-SD complexes in Emx1-Kcnq2 cKO and Scn8a+/N1768D GOF mouse cortex. In these models, SD appears following seizure activity simultaneously in bilateral anterior and posterior electrodes. (E) Recurrent SD-like events during status epilepticus. In a peritumoral epilepsy model, multiple SD-like events (red asterisks) appear during the early phase of unabated status epilepticus (unpublished data). Inset shows expanded trace of the first SD-like event appearing during the onset of the status epilepticus. Note seizure activity overriding the DC potential shift and its transient disappearance after repolarization. SD: spreading depolarization.
SD and Acquired Epileptogenesis
SD commonly follows acute brain injury and its incidence may associate with subsequent epilepsy. Like seizures, SD leaves enduring transcriptomic changes including upregulation of immediate early genes and growth factors, induction of long-term synaptic potentiation,19-21 and initiation of epileptic discharges in epileptic brain. 22 An unusual cogeneration pattern of seizures and SD (“spreading convulsion”) has been detected in subarachnoid hemorrhage, 23 and the occurrence of post-injury SD showed a trend toward subsequent epileptogenesis. 24 Longitudinal clinical studies with SD detection are rare and it remains unclear whether SD acts as a driver of epileptogenesis or simply reflects the severity of the acute injury.
While the contribution of SD to human epileptogenesis remains to be clarified, SD has been detected in brains undergoing experimental epileptogenesis. In kindling models, recurrent seizure-SD complexes arise within toxin-injected hippocampus during the epileptic phase. 25 Recurrent SD waves are also seen during early tumorigenesis in a mouse glioblastoma model. At the initial stage of neocortical tumor invasion, once epileptiform spiking has begun, isolated spontaneous cortical SD waves emanate from the leading tumor edge shortly before the onset of spontaneous seizures. 26 In this model, SD appears during the transition from the pre-ictal to the ictal stage, thus marking the “end of the beginning” of epileptogenesis. However, SD is not the sole contributing factor; at this stage, the tumor edge displays striking elevation of extracellular glutamate 27 and pathogenic transcriptome changes in known human epilepsy genes that favor hyperexcitability. 28
Like Seizures, In Vivo SD Thresholds are Dynamic
SD susceptibility is not fixed and depends on circuit pathophysiology, arousal level, circadian phase, hormonal cycles, temperature, and cerebral metabolism. Just as human seizure disorders show individualized dependence on these key variables, 29 chronic DC monitoring has identified prominent gene-specific diurnal SD patterns in mouse models, eg, higher during light phase (Kcna1 KO, Scn8aN1768D/+),17,18 dark phase (Emx1-Kcnq2 cKO, ATP1A2T345A),12,17 and during the transition of light/dark cycle (Scn1a+/R1407X). 10 Gene-specific patterns suggest that the underlying network instability responds differently to intrinsic or extrinsic (eg, cyclic autonomic and hormonal activity) modulation that contributes to threshold variability in the susceptible brain. In brain-injured patients, brain temperature, hypotension, and a spike of intracranial pressure correlate with SD susceptibility,30-32 while their relevance to SD occurrence in atraumatic human epileptic brain is unknown. A past history of seizures or SD can also affect subsequent SD susceptibility. For example, recurrent seizures cumulatively render brain tissue resistant to SD 33,34 and daily SD induction incrementally increases SD threshold. 35
End of the Beginning: is SD an Innate Brain Mechanism Limiting a Seizure?
In the absence of any unifying cellular mechanism that terminates seizures, a recent report 36 raises an intriguing hypothesis that a seizure-triggered SD might serve as this intrinsic mechanism. In that study, convulsant-evoked seizures in anesthetized mice were transiently interrupted by spontaneous or triggered SD, creating a minutes-long seizure-free refractory period. Prior SD induction also reduced ictal activity in response to subsequent convulsant challenges. These results support an anti-seizure effect of SD in anesthetized wild-type mouse brain.
However, experiments in unanesthetized genetic models with spontaneous SD generation tell a different story. In Kcna1 and Scn1a mutant mice, most spontaneous seizures self-terminate without SD, and even when postictal SD is present, it may appear many seconds to minutes after full termination of the electrographic seizure (Figure 1B).
A second issue is whether SD suppresses future seizure activity. While not common, clinical intracranial recordings have detected seizures generated immediately following SD, and even seizure activity that overrides the repolarization phase of SD and continues for minutes.23,37 We detected post-SD seizures in a recent study in Scn8aN1768D/+ GOF epilepsy mice, where a clinically robust, tonic motor seizure appeared within a minute following a seizure-SD complex in one-third of spontaneous events. 18 In the brain tumor mouse model, 26 recurrent SD-like DC-potential shifts were detected at the beginning of hours-long ictal activity or status epilepticus (Figure 1E, our unpublished observation). In this model, SD or SD-like events transiently reduced ictal activity for tens of seconds, but over a longer time window recurrent SD actually associated with the initiation of prolonged seizures. Such “SD-seizure” sequences are not well studied but have been reproduced when SD is triggered in preconditioned neuronal circuits/cells to enhance excitability.38,39 Thus, unlike in healthy brain tissue, SD can (paradoxically) represent an innate pro-convulsive mechanism when the neural circuit is predisposed to hyperexcitability.
Does SD Underlie the Postictal Generalized EEG Suppression?
So if the answer to the question “Why do seizures stop?” is not “because they trigger an SD wave,” could SD underlie postictal decrements in EEG amplitude? This phenomenon, postictal generalized EEG suppression (PGES), is a frequent terminal feature of generalized tonic seizures, with a suggestion, still in debate, that it might serve as a biomarker of sudden death in epilepsy (SUDEP) risk. 40 Human scalp-recordings show simultaneous onset of a period of low amplitude EEG across all electrodes that could reflect an underlying SD event; however, there is no evidence of sequential appearance in neighboring electrodes.
The cellular basis for PGES remains unknown; however, we find little supporting evidence for this proposal in mouse models. In mouse intracranial DC recordings, we detected generalized tonic clonic seizures terminating with a striking PGES-like period of flattened ECoG amplitude but no evidence of DC shift (Figure 2). When an SD does occur, the onset is about 1 min after the PGES pattern has ended.
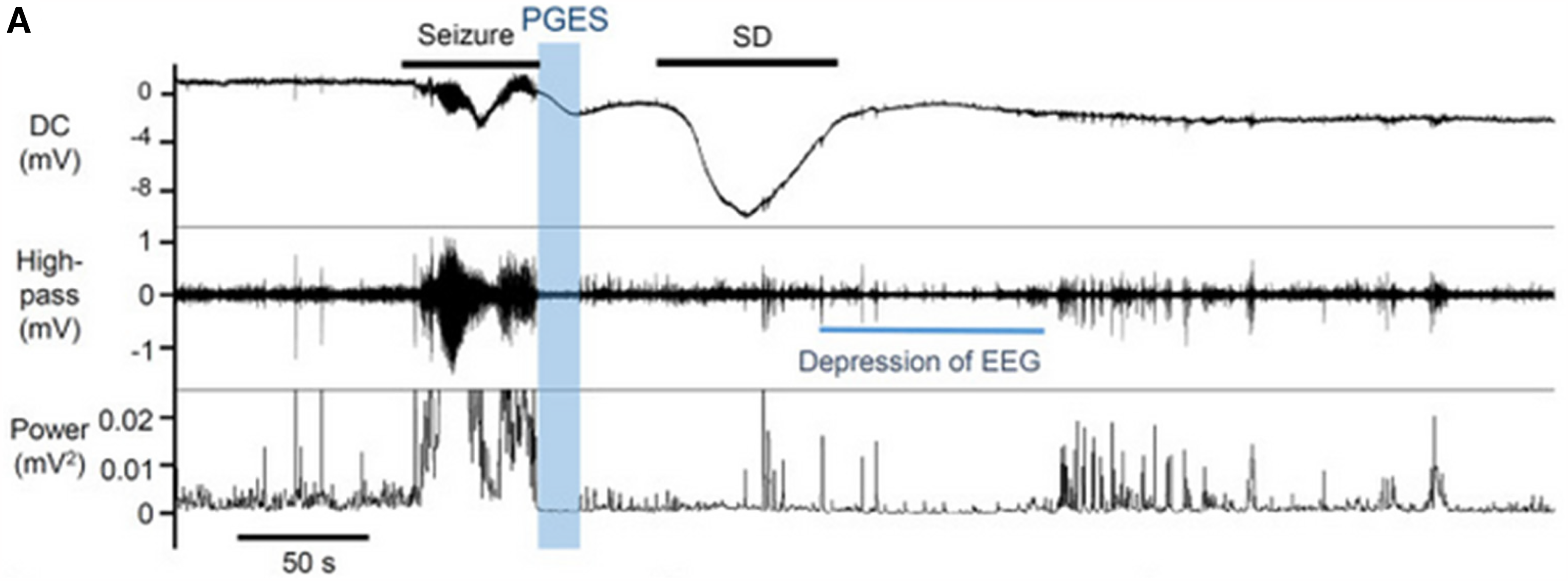
Postictal generalized EEG suppression (PGES) is distinct from SD EEG depression. A seizure-SD complex interleaved by an instantaneous PGES in an awake Scn1aR1407X/+ mouse. The generalized seizure was terminated with PGES and an SD followed with a minute delay. Top: DC, middle high-pass filtered (>1 Hz), bottom EEG power. Data from Aiba 2023. 10 SD: spreading depolarization.
Beginning of the End: the Lethal Brainstem SD-SUDEP Correlation
Analysis of SUDEP has identified additional SD-related genes that may presage a highly consequential impact of postictal SD, namely, a terminal seizure that ends life. The initial hypothesis that a wave of SD invading brainstem cardiorespiratory circuitry following a seizure could provoke sudden death arose from the analysis of SD in genetic mouse SUDEP models.41,42 These studies demonstrated that epileptogenic ion channel mutations lower the threshold for postictal SD in the dorsal medulla following a cortical seizure and showed that the resulting wave of brainstem depolarization could depress cardiorespiratory pacemaking and cortical EEG on a timespan consistent with the postictal aftermath of apnea, bradycardia, and asystoles characteristic of lethal seizures in patients. 43 Subsequent reports have identified brainstem SD in other mouse models with early mortality.44-46 Despite the growing evidence for lethal effect of SD when it invades the lower brainstem in mice, there is as yet no direct human evidence for this candidate SUDEP mechanism.
Conclusion
Spontaneous episodes of cortical SD are a prominent electrical phenotype in mouse epilepsy models, in some cases appearing earlier and outnumbering seizures themselves. The search for genetic and cellular mechanisms underlying seizure and SD thresholds and the impact of repeated SD episodes on neural circuitry are an integral part of epilepsy research, and future discovery of a means of reducing their co-occurrence may have life-saving consequences.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institute of Neurological Disorders and Stroke, Blue Bird Circle Foundation, (grant number R0129709).