
Abstract
Genetic epilepsies and developmental and epileptic encephalopathies are commonly associated with concomitant movement disorders, which can mimic seizures and/or create additional disability. Appropriate diagnosis is critical to proper management. A broad range of movement disorder phenomenologies occur among patients with genetic epilepsy, including dystonia, chorea, ataxia, myoclonus, stereotypy, tics, and Parkinsonism. For some genes, clear relationships exist between genotype and movement disorder phenotype, while in other situations, the relationship is more complex. Diagnosis of movement disorders involves elements of history, physical examination, video review, and neurophysiology. Assessment of associated impairment, distress, and/or safety concerns is important for considering risks/benefits of treatment. Movement disorders may range from severe and dangerous, requiring pharmacologic or neuromodulatory treatments such as deep brain stimulation, to relatively benign, with only reassurance and continued observation required. Appropriate treatments differ based on phenomenology and etiology, with some genes associated with tailored treatments which can provide dramatic benefit.
Introduction
Genetic epilepsies, frequently childhood onset, are often associated with additional neurodevelopmental symptoms beyond seizures. Concurrent movement disorders are common among patients with genetic epilepsy, particularly among those with developmental and epileptic encephalopathies (DEEs). 1 Distinguishing between seizure and movement disorder in these patients is important to optimize treatment and prevent unnecessary side effects.
Several genes are associated with both epilepsy and movement disorders, with some patients presenting with epilepsy, some patients presenting with movement disorder, and some patients with both. This phenotypic variability relates to genotype in some cases and is more complex in others.
Both the clinical presentation and the underlying etiology may influence the appropriate treatment of movement disorders associated with epilepsy. Certain disorders have etiology-specific treatment, such as the ketogenic diet in GLUT1 deficiency syndrome. In other situations, impact on life and phenomenology guide treatment decisions.
In this narrative review with several genes as case examples, we will describe common movement disorder phenomenologies, list commonly reported epilepsy and movement disorder phenotypes associated with genetic epilepsy by gene, consider the importance of genotype–phenotype associations, and review considerations for diagnosis and treatment.
Clinical Characteristics
Movement disorders are typically characterized by phenomenology, as this can inform understanding of localization and etiology and guide treatment options. Broadly speaking, movement disorder phenomenologies can be characterized as hyperkinetic, ataxic, or hypokinetic. Hyperkinetic phenomenologies are further differentiated by suppressibility, stereotyped versus patterned, posturing versus random characteristics of movement, and influence of voluntary movement on severity of movements. Movement disorders may be continuous or paroxysmal, with paroxysms frequently, but not always, associated with provoking factors, such as in the case of kinesiogenic and/or exertional dyskinesias.
Definitions and descriptions of phenomenology of hyperkinetic or ataxic movements in the pediatric population including dystonia, chorea, athetosis, myoclonus, tremor, tics, stereotypy, and ataxia have been developed and detailed elsewhere.2–4 We will review key features, helpful for differentiating these phenomenologies on exam, here.
Dystonia involves sustained or intermittent and/or jerky contractions, which can involve twisting or posturing, co-contraction of agonist–antagonist muscles. Dystonia is more variable than spasticity, and is typically worsened by voluntary movement and/or emotion.
Chorea involves ongoing random discrete involuntary movements which are often described as “dance-like” while athetosis involves more slow writhing movements. Both are associated with motor impersistence and typically worsen with voluntary movement and/or emotion.
Myoclonus involves brief lightning-like jerks due to sudden muscle contraction which are often repeated but non-rhythmic.
Tremor is a rhythmic oscillatory movement about a null-point.
Tics and stereotypies are brief, suppressible, stereotyped movements. Tics are typically associated with an urge, which young and/or nonverbal children may not be able to describe, and may be briefly suppressible. Stereotypies are repetitive movements that are often associated with excitement or emotion. Stereotypies generally begin before age three and maintain the same pattern over time while tics usually begin at later ages with a waxing and waning pattern that involves shifts in the patterned movement or sound observed.
Ataxia, a disorder of coordination rather than excessive involuntary movement, involves inability to generate an expected voluntary movement trajectory, not attributed to weakness or involuntary muscle activity. Ataxia is frequently associated with wide-based gait and/or difficulty with tandem gait, dysmetria of limbs and eyes, and oculomotor apraxia.
Parkinsonism, relatively infrequent in the pediatric population, involves resting tremor, bradykinesia (slowness of movements with decrement in amplitude of movement over time), rigidity (increased tone without velocity dependence of spasticity), and postural instability, when described in adults. Many of these features are also described in the pediatric population. 5
These terms have been used to characterize phenomenology of movement disorders commonly reported in patients with genetic epileptic disorders, reported in association with genes and epilepsy phenotype in Table 1. Additional movement disorder phenomenology terminology in this chart includes dyskinesia, a general term often used when overlap between dystonia, chorea, and/or athetosis occurs, ballismus, with involves large-amplitude choreiform movements, and paroxysmal tonic upgaze, a brief dystonic involuntary eye movement.
List of Genes Commonly Reported in Association With Both Epilepsy and Movement Disorders.
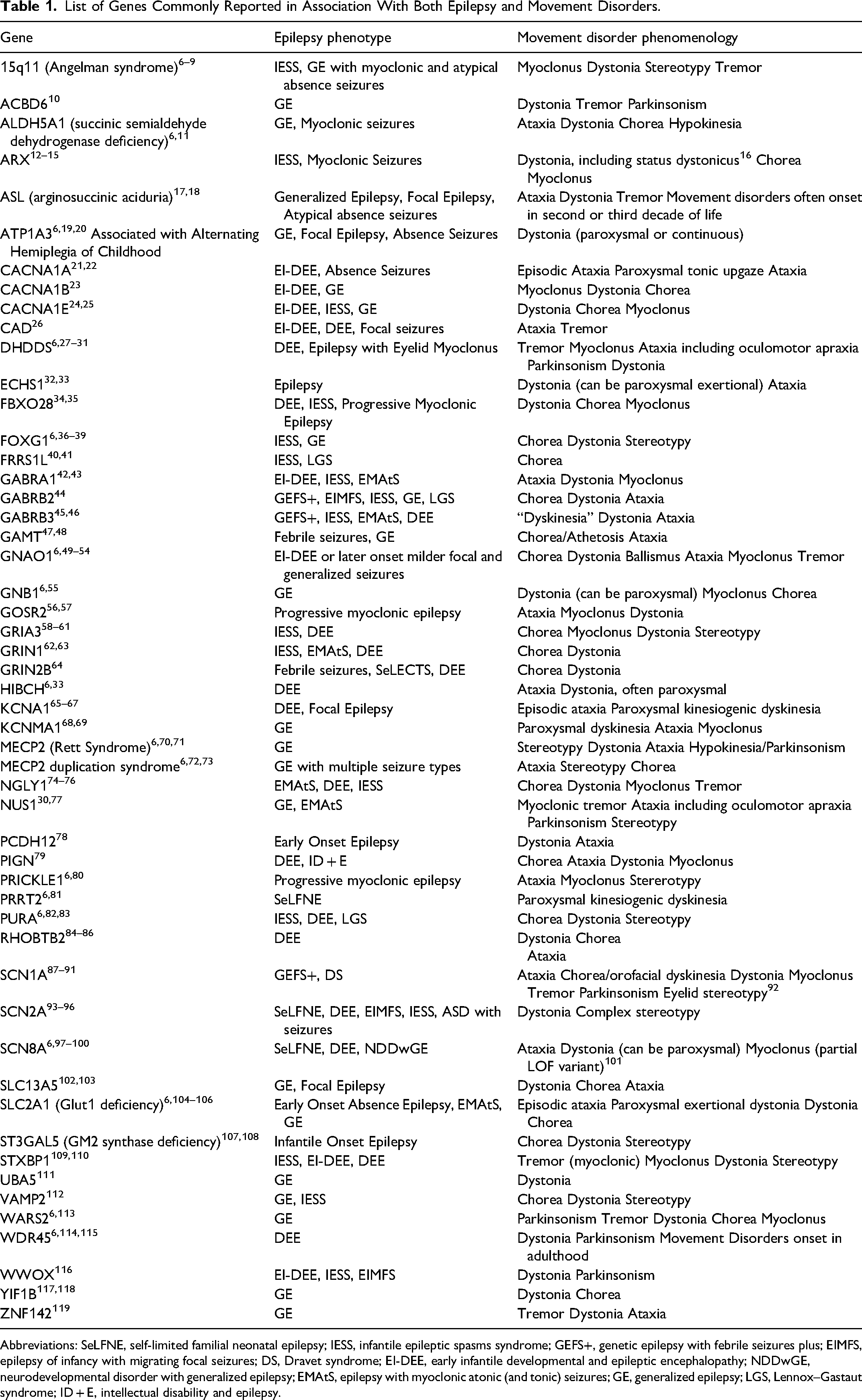
Abbreviations: SeLFNE, self-limited familial neonatal epilepsy; IESS, infantile epileptic spasms syndrome; GEFS+, genetic epilepsy with febrile seizures plus; EIMFS, epilepsy of infancy with migrating focal seizures; DS, Dravet syndrome; EI-DEE, early infantile developmental and epileptic encephalopathy; NDDwGE, neurodevelopmental disorder with generalized epilepsy; EMAtS, epilepsy with myoclonic atonic (and tonic) seizures; GE, generalized epilepsy; LGS, Lennox–Gastaut syndrome; ID + E, intellectual disability and epilepsy.
Movement disorders can frequently be disabling, given their common interference with voluntary movement, potential for discomfort, and in severe cases, potential for injury and/or rhabdomyolysis. However, it is worth noting that movement disorders can also frequently be noticeable without being impairing or distressing. This is particularly the case for stereotypy and often for tics, but can be true of any phenomenology. Therefore, clinical evaluation of movement disorders requires both identification of phenomenology and evaluation of associated disability and distress, if any.
Genotype–Phenotype Associations
Given the clinical heterogeneity in many disorders with concomitant movement disorders and epilepsy, attention to genotype–phenotype correlations may inform clinical treatment. Increasingly, specific phenotypic patterns are related to specific variants which can have implications for treatment choices and prognostication.
For example, in SCN8A, both epilepsy and movement disorder phenotypes are associated with the genotype and functional impact. Gain-of-function (GoF) variants are frequently associated with a severe epilepsy phenotype with focal-onset seizures in infancy, while loss-of-function (LoF) variants are associated with intellectual disability without epilepsy as well as epilepsy manifest as generalized, primarily absence, seizures with onset after infancy.120–122 While movement disorders can occur in both GoF and LoF variants, chronic ataxia and myoclonus are particularly described in LOF variants.98,101 Sodium-channel blockers, particularly useful in SCN8A-related GoF disorders, are not generally effective with SCN8A LoF variants. 123
GNAO1-related disorders are exemplary of the clinical implications and complexity in this area. Since the first description in 2013, about 200 variants have been associated with a spectrum of movement disorders and epileptic encephalopathy, as well as variable overlap. Nearly half of the reported variants are in a handful of “hot spots,” or recurrent variants, and correlate to a particular phenotype. 124 For example, the most commonly reported variants at the p.R209 residue typically display a severe movement disorder with dystonia, chorea and hyperkinetic crises with a milder, non-DEE epilepsy. In contrast, variants in the p.G40 residue commonly have DEE with a milder movement disorder. While initially a dichotomous mechanism was proposed in which GoF variants were associated with movement disorders and LoF associated with epileptic encephalopathies based on a cAMP assay of downstream GTPase activity variants, 50 this did not fully explain the clinical heterogeneity. A third hot spot, p.G203R typically manifests as DEE and a movement disorder with hyperkinetic crises. Recently, milder later-onset dystonia, including inherited cases, have been associated with novel GNAO1 variants and presumed to be due to haploinsufficiency. 125 The full understanding of this heterogeneity in g-protein signaling remains incompletely understood.
Other examples of disorders in which genotype influences movement disorder phenotype include SCN1A, where GoF mutations are more likely to be associated with movement disorders, 87 KCNMA1, where GoF mutations often present with paroxysmal dyskinesia while LoF variants often are associated with other phenemonologies, 69 MECP2-related disorder (Rett syndrome) where truncating mutations are associated with more severe dystonia and rigid-akinetic syndrome, 70 and GRIA3 where GoF variants are more likely to be associated with movement disorders, and earlier onset of epilepsy. 60
By studying these correlations, clinicians can better predict disease progression, identify at-risk individuals, and tailor interventions to the unique needs of each patient, advancing personalized medicine and improving outcomes.
Diagnosis
Distinguishing movement disorders and seizures often presents a challenge in patients with epilepsy and DEEs, particularly when there is overlap in both the phenomenology and underlying genetic cause. Since genetic variants co-segregate in families presenting with diverse epilepsies and movement disorders with significant phenotypic hetereogenity, 126 we cannot rely on genotype alone to differentiate seizures from movement disorders. This distinction is important: misclassification of seizures and movements impacts patient care and may affect clinical trial outcomes. 127 Treatment for seizures and movement disorders are often vastly different.
The first step in evaluating motor symptoms in a patient with genetic epilepsy involves a history and neurologic examination, which in many cases is sufficient to identify features of specific movement disorder phenomenologies as detailed above. Assessing the influence of voluntary movement on the involuntary movement can help distinguish between movement disorders and seizure, such as with exacerbation of the involuntary movement in the case of dystonia or chorea and suppressibility with tics or stereotypy. Some paroxysmal disorders may be provoked in clinic for direct observation, such as using excitement to provoke a stereotypy or sudden movement after a prolonged rest to provoke paroxysmal kinesiogenic dyskinesia. Video recording the examination can allow for more careful evaluation for these features. However, a lack of diagnostic clarity may occur when paroxysmal movements (a) may not be observed during a clinical encounter and (b) may have a clear onset/offset which raises concern for an epileptic etiology. Some phenomenologies, such a myoclonus, could have an epileptic etiology and require further evaluation, and some movement disorder presentations may raise concern about atypical features, such as an apparent tic or stereotypy which does not appear to be suppressible.
In these situations, video EEG is the gold standard for distinguishing epileptic from non-epileptic events including movement disorders. It should be noted that there are many other etiologies of paroxysmal non-epileptic events apart from movement disorder. Many of these are specifically described in genetic epilepsies and DEEs, including neuro-irritability related to internal or external stimuli, autonomic dysfunction, severe behavioral disturbances, and catatonia. 128 Therefore, the absence of epileptic findings on EEG is not sufficient to diagnose movement disorder as the etiology of a paroxysmal event.
Further complicating EEG interpretation is the observation that only 21% of focal aware (formerly “simple partial”) seizures have ictal EEG change on scalp EEG. 129 Synchronized cortical activity from a minimum of 6 cm2 and, more typically 20 cm2, is required to produce a spike on scalp EEG. 130 Therefore, a “negative” ictal scalp EEG does not exclude seizures. This highlights the importance of also considering history and physical examination in distinguishing seizures from movement disorders and other types of spells. Videos of events, now commonplace with smartphones, are also fairly accurate when viewed by trained experts. 131
Additional neurophysiologic evaluations, such as surface electromyography (EMG), can be helpful for distinguishing movement disorder phenomenologies, sometimes in combination with surface EEG. In the case of surface EMG, electrodes are placed on muscles involved in the abnormal movements, ideally including agonist/antagonist muscle pairs on both sides of the body to allow for evaluation of synchrony between limbs and coherence between agonist/antagonist muscles. Methods for acquiring and evaluating this data are well detailed by Hallett et al 132 Surface EMG synchronized with video can allow for objective evaluation of exacerbation and/or suppression of involuntary movements with voluntary movement or other stimuli. It can also be a useful adjunct in distinguishing between phenomenologies and localizing the neuroanatomical origin of myoclonus.
Considering the special case of myoclonus, distinguishing origin of the myoclonus is challenging and frequently requires adjunctive neurophysiologic studies. For epileptic myoclonus, this is typically (but not always) associated with spikes or polyspikes on scalp EEG, synchronized with short burst duration (<50 ms) surface EMG activity. Epilepsia partialis continua is an exception as it can be EEG-negative given highly focal origin and involves burst duration which is often longer than 50 ms. Particularly in DEEs, careful consideration of epileptic myoclonus is indicated for patients with semi-rhythmic myoclonus which can clinically resemble tremor. 6
Non-epileptic cortical myoclonus may be diagnosed by identifying a cortical potential prior to myoclonus onset using jerk-locked back-averaging of the EEG. Subcortical origins of myoclonus, such as thalamus-basal ganglia, reticular, propriospinal, spinal, or peripheral, carry their own neurophysiologic signature whose identification can help guide treatment choices. 7
While less studied than myoclonus, other types of movement disorders can be distinguished by neurophysiologic features, such as via surface EMG. For example, dystonia involves prolonged bursts of muscle activity which may be tonic, phasic, or rhythmic and abnormal activation of agonist/antagonist muscles (co-contraction) or contralateral muscles (overflow). 133
Finally, it should be noted that as patients with genetic epilepsies can have concurrent movement disorders and seizures, the identification of a movement disorder does not exclude the potential for seizure and vice-versa, as many patients will have both, potentially presenting at different ages.
Treatment
Treatment of movement disorders in DEEs is often guided by phenomenology, but in some circumstances, treatment can be more precision-based when there are known interventions for a specific disorder or genotype. Not all movement disorders require treatment; less than half of patients in a recently-described cohort required treatment for their movement symptoms. 1 For many movement disorders, the risks and potential side effects of treatment may outweigh the benefits, and each family may have different preferences and values regarding the risks and benefits of treatment. Pharmacotherapy can be considered when the movements cause distress or interrupt function, or if there is potential for harm from the movements. Stereotypies, for example, are generally not harmful and so do not typically require treatment. In rare circumstances where stereotypies pose risk of injury or are otherwise disruptive, behavioral treatment is the first-line approach, 134 but may be of limited utility for individuals with DEE due to associated developmental delay or cognitive impairment. Some additional movement phenotypes may also lack effective treatments. For example, there are no approved symptomatic therapies for ataxia, though 4-aminopyridine has shown benefit in spinocerebellar ataxia and KCNA1 cohorts.135,136 Overall, these studies have small sample sizes and it is hard to generalize these results to other causes of ataxia.
Chorea, dystonia, and myoclonus are hyperkinetic movement disorders that are more likely to require treatment. Common pharmacologic agents used for dystonia include levodopa, anticholinergic agents such as trihexyphenidyl, benzodiazepines, baclofen, clonidine, or VMAT2 inhibitors such as tetrabenazine. Chemodenervation is also effective for treating dystonia, but best suited for focal dystonia or targeting a more problematic body region. Treatment options for chorea include dopamine blockade with typical or atypical antipsychotics, or dopamine depletion with VMAT2 inhibitors. For severe hyperkinesis, deep brain stimulation, particularly in individuals with GNAO1, has been helpful in preventing further hyperkinetic crises. 137 DBS has been used as rescue for status dystonicus with benefit for hyperkinetic movement disorders associated with other genetic epilepsies, including SCN2A, UBA5, and GNB1.55,96,138 Non-epileptic myoclonus is often treated with anti-seizure medications similar to those used in myoclonic epilepsies; levetiracetam, valproate, and benzodiazepines are often implemented. However, efficacy may vary based on the localization of the myoclonus (cortical vs subcortical vs segmental). 139
Paroxysmal dyskinesias, although still consisting of dystonia, chorea, and ataxia in isolation or combination, may differ in their treatment compared to the more continuous movement phenotypes discussed above. Paroxysmal kinesigenic dyskinesia (PKD), often associated with variants in PRRT2, usually responds extremely well to very low doses of sodium channel blockers. Paroxysmal movement disorders in RHOBTB2 are also reported to respond to sodium channel blockers. 85 Paroxysmal exercise-induced dyskinesia (PED), which can be phenotypically differentiated from PKD in that more sustained exercise induces the abnormal movements rather than onset of exercise, is associated with GLUT1 deficiency syndrome and variants in SLC2A1. First-line treatment of PED associated with GLUT1 is with use of ketogenic diet. Paroxysmal non-kinesigenic dyskinesia (PNKD) episodes due to the N999S variant in KCNMA1 are often triggered by excitement (as in cataplexy) or tactile stimuli and exhibit a remarkable response to lisdexamfetamine. 140 In episodic ataxia due to variants in KCNA1 (EA1) or CACNA1A (EA2), episodes frequency or intensity can be reduced with carbonic anhydrase inhibitors such as acetazolamide.
Additionally, many inborn errors of metabolism are associated with both epilepsy and movement disorder. Dietary and/or gene therapies which address underlying metabolic deficient and/or toxic accumulations can also reduce movement disorder and epilepsy symptoms. Examples from genes listed in Table 1 include GAMT, where both epilepsy and movement disorders improve with creatine supplementation, 47 and CAD, which is treatable with uridine supplementation. 26
Discussion
The approach to the patient with concomitant epilepsy and movement disorder from a genetic disorder is challenging, but an evaluation that jointly considers phenomenology, impact, and etiology of a patient's movement disorder is essential to prevent inappropriate or unnecessary treatment and treat disabling symptoms in a tailored manner. Evaluation for movement disorders in genetic epilepsy should extend beyond consideration of movement disorder in the differential diagnosis for paroxysmal spells and include physical examination for continuous movement disorders such as dystonia or chorea which may impact functional abilities, comfort, and/or safety. In some situations, etiology-specific treatment targeted to underlying genetic diagnosis can impact both seizures and movement disorders. We provide a summary of our recommended approach to possible movement disorders in a patient with genetic epilepsy in Figure 1.
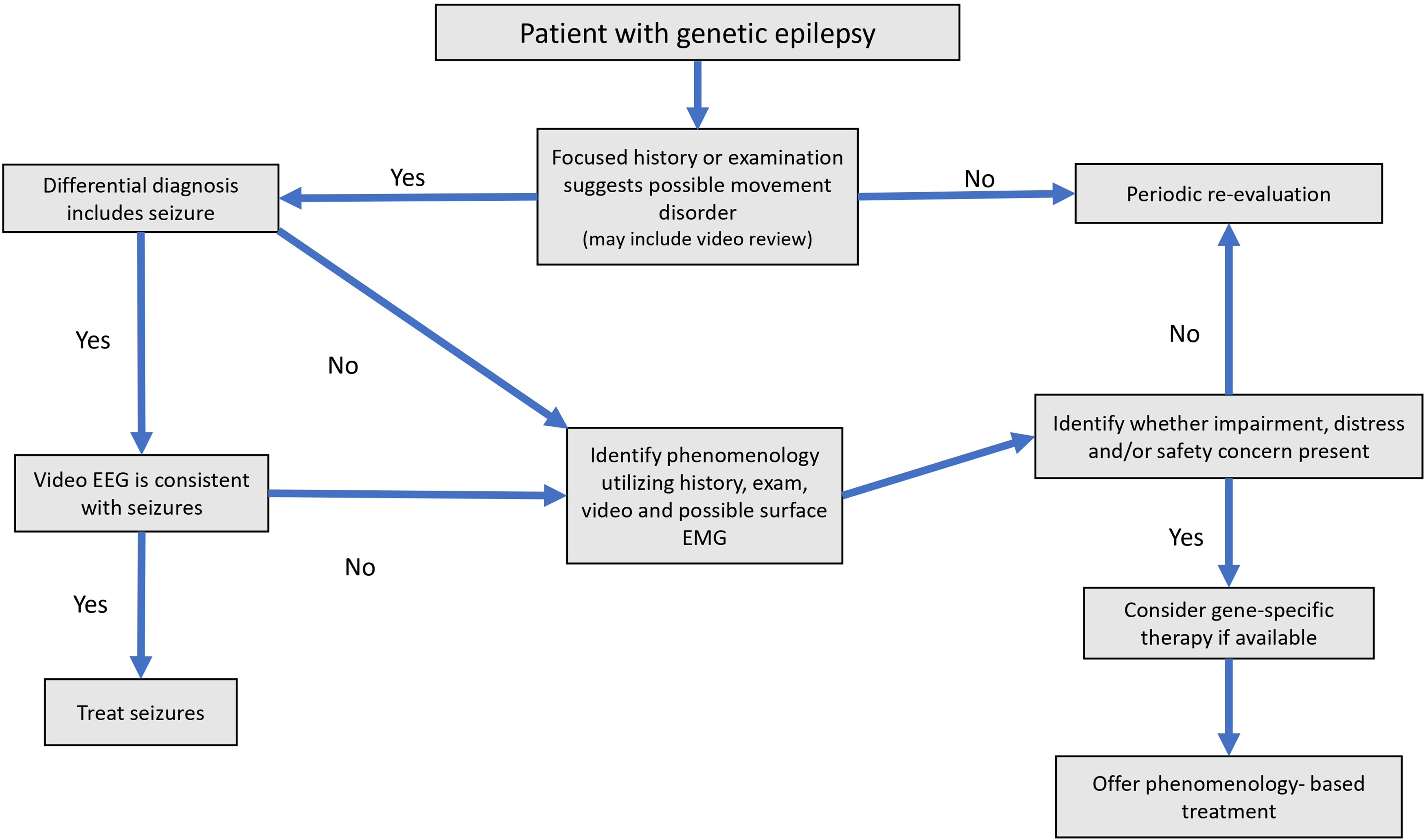
Suggested approach to patient with genetic epilepsy to evaluate for possible concomitant movement disorder.
This review and the medical literature overall have several limitations regarding this broad topic. Descriptions of movement disorders associated with epilepsy as reported in the literature may skew towards more severe phenotypes. For example, we found that stereotypies were infrequently mentioned as movement disorders associated with genetic epilepsy, but when a cohort of patients with genetic DEEs were evaluated for movement disorder characterization, stereotypy was the most commonly identified phenomenology. 1 Poor ascertainment likely results in under-reporting of movement disorders in these patients. Additionally, inconsistent and overlapping terminology can create challenges in a broader understanding of this topic. Frequently, subjectivity is required for distinguishing movement disorder phenotypes and/or treatment response in clinical practice, which can also limit scholarly descriptions.
Conclusion
Movement disorders are commonly associated with genetic epilepsy and require proper identification and a tailored diagnostic and therapeutic approach is needed to provide best patient care.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.