
Abstract
In recent years, there has been a significant increase in preclinical studies to test genetic therapies for epilepsy. Some of these therapies have advanced to clinical trials and are being tested in patients with monogenetic or focal refractory epilepsy. This article provides an overview of the current state of preclinical studies that show potential for clinical translation. Specifically, we focus on genetic therapies that have demonstrated a clear effect on seizures in animal models and have the potential to be translated to clinical settings. Both therapies targeting the cause of the disease and those that treat symptoms are discussed. We believe that the next few years will be crucial in determining the potential of genetic therapies for treating patients with epilepsy.
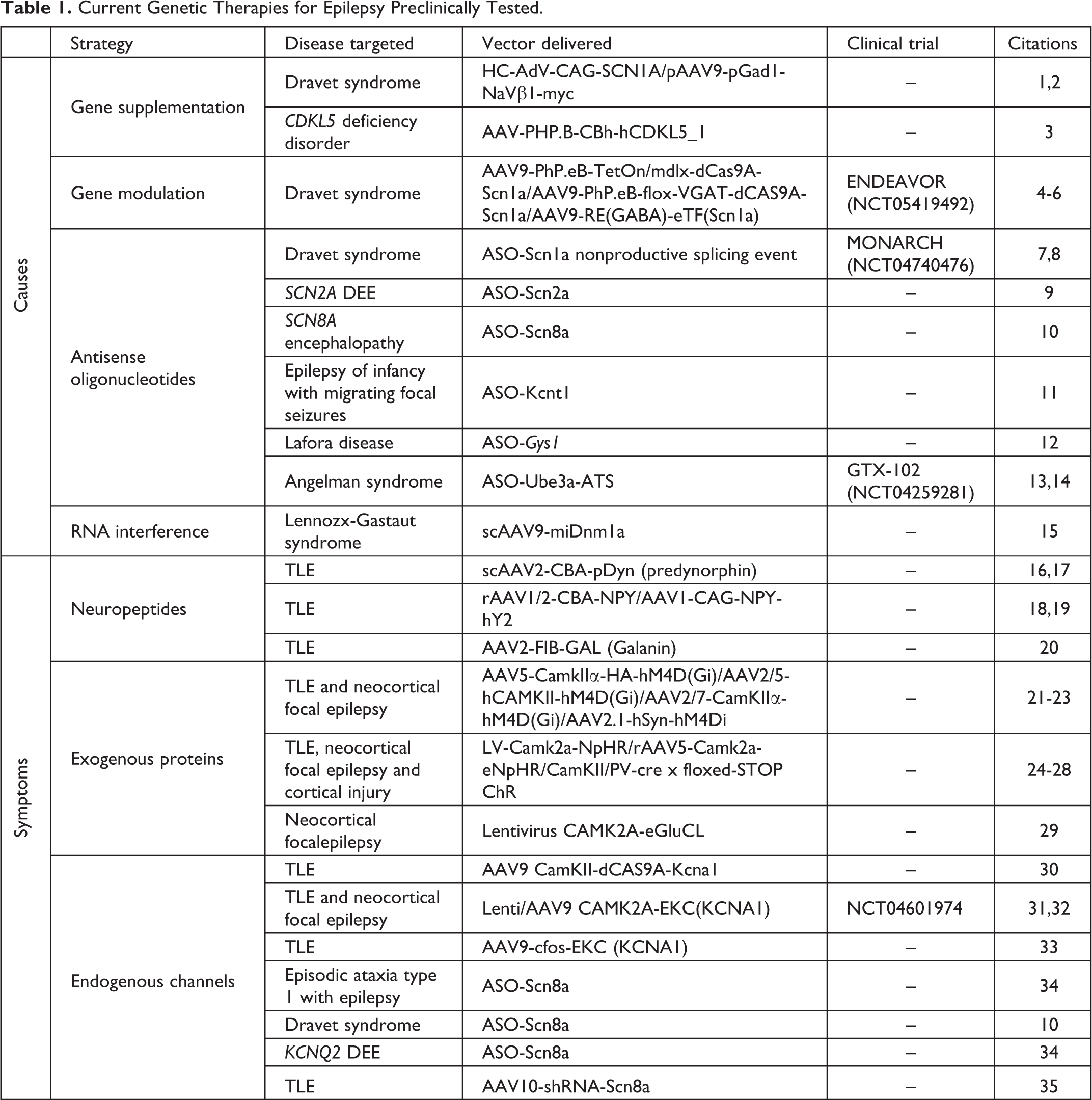
Epilepsy is one of the most common severe neurological diseases with many patients having refractory seizures without any effective options for treatment. Recently, several genetic therapies have been developed with the aim of treating this intractable population of patients. Among these approaches some are already in or nearing clinical trials, bringing hope to the epilepsy field and to the patient community. These treatments can be divided in two broad categories: genetic therapies targeting the cause of the epileptic phenotype, and those aiming to address the symptoms, namely the seizures (Table 1).
Current Genetic Therapies for Epilepsy Preclinically Tested.
Treating the Cause
Although epilepsy overall shows strong heritability, there are several, individually rare, monogenic syndromes. These are commonly de novo, although can also be inherited in a dominant or recessive manner. Among causative genes are several ion channels (SCN1/2/8A, KNCA1, KCNQ2/3, GABRA1, KCNT1), mutations of which are associated with the umbrella class of developmental and epileptic encephalopathies (DEEs). The genetic background of DEEs and their resistance to conventional anti-seizure therapies identify these diseases as obvious candidates for genetic therapy. By modifying the expression or activity of the implicated gene products, the goal of such therapies is to correct the underlying molecular pathology. 36
Gene Supplementation
Loss-of-function mutations (LoF) are in principle treatable with gene supplementation of a healthy copy of the mutated gene, for instance using a viral vector. Indeed, gene supplementation has been successfully trialed in mouse models of Cyclin-dependent kinase-like 5 (CDLK5)-deficiency disorder, an X-linked DEE caused by de novo LoF mutations in the CDKL5 gene. Gene supplementation with a human CDKL5, delivered with an adeno-associated viral vector (AAV), successfully ameliorated the disease phenotype of Cdlk5 knock-out mice. 3 Recent work has also demonstrated the viability of gene supplementation to improve outcomes in mouse models of Dravet syndrome, a genetic DEE frequently caused by haploinsufficiency of the voltage-gated sodium ion channel Nav1.1 secondary to de novo mutations in SCN1A. Adenovirus (AdV)-mediated delivery of SCN1A reduces spontaneous seizures and mortality in Dravet syndrome mouse models, as does AAV-mediated delivery of SCN1B (the beta-subunit of Nav1.1, which is smaller and has been shown to promote trafficking of alpha subunits to the cell surface). 1,2 Although gene supplementation is a straightforward approach, it is limited by several considerations: AAV vectors have a restricted packaging capacity, which is exceeded by the full-length SCN1A open reading frame; conversely, AdV vectors do not spread widely in the central nervous system (CNS) and are potentially toxic; finally, it is unclear which neurons and which brain regions need to be treated, and at what developmental stage, in order to achieve an optimal therapeutic effect.
Gene Modulation
Ion channels are among the most important genes implicated in neurodevelopmental epilepsies but are often too large for straightforward gene supplementation. To overcome this obstacle, a modified version of the gene editing tool CRISPR/Cas9, known as CRISPR activation (CRISPRa), has shown potential in treating Dravet syndrome. Using CRISPRa targeting of the SCN1A promoter, it is possible to upregulate the expression of SCN1A. Accordingly, a recent paper has demonstrated that AAV delivery of CRISPRa constructs into a mouse model of Dravet syndrome increases Nav1.1 protein levels, rescues excitability of cortical parvalbumin-positive (PV+) neurons, and reduces susceptibility to thermally induced seizures otherwise observed in the model. 4 Another study achieved a therapeutic effect in a mouse model of Dravet syndrome crossed into a mouse line constitutively expressing most of the CRISPRa machinery. 5 In both of these studies, the transcriptional upregulation was biased to inhibitory interneurons, which fail to tolerate SCN1A haploinsufficiency, and the anti-epileptic effect is thought to be mediated by an increase in their excitability.
Interestingly, a similar approach using defective Zinc Finger Nucleases fused to a transcriptional activator targeted to the Scn1a promoter has also been shown to be effective in a mouse model of Dravet syndrome. Another study achieved upregulation of SCN1A in inhibitory interneurons using an AAV to express a GABArgic cell-selective transcription factor, resulting to an increase in Nav1.1 protein expression. 6 Additional safety studies in nonhuman primates (NHPs) showed good uptake of the construct throughout the brain and with no adverse events. 6 In principle, these approaches can be used to upregulate any gene, and thus treat a range of LoF mutations causing epilepsy, although implementation requires substantial bioinformatic insights into promoter and enhance function.
Antisense Oligonucleotides and RNA Interference
Several de novo mutations causing epilepsy in ion channels are gain-of-function (GoF) and so are not amenable to gene supplementation. For these mutations decreasing expression at the gene or messenger RNA (mRNA) level could potentially have a therapeutic role. Antisense oligonucleotides (ASOs) are short single-stranded nucleic acid sequences, typically modified to increase their stability, which are capable of targeting an mRNA transcript of interest to modulate pre-mRNA splicing events, or regulate translation, thus modulating the efficiency of transcript processing and the production of a protein of interest. In recent years, ASOs have been shown to ameliorate epileptic phenotypes in murine models of several GoF DEEs. Scn2a ASOs have been administered to Scn2a mutant mice, reducing seizure frequency, and extending life span. 9 Similarly, reduction of Scn8a transcripts using ASOs has been shown to improve the frequency of behavioral and electroencephalographic seizures and overall survival of Scn8a mutant mice. 10 Antisense oligonucleotides therapy has further been shown to improve outcomes in mouse models of other epilepsy-associated syndromes, including KCNT1-associated epilepsy of infancy with migrating focal seizures, 11 myoclonic epilepsies such as Lafora disease, 12 and Angelman Syndrome. 13,14 It has also been shown to reduce aberrant transcript expression in patient-derived fibroblasts in Unverricht-Lundborg disease. 37
Importantly, while ASOs typically knock-down transcript activity, they also have potential for upregulating gene expression, making them suitable for use in LoF genetic variants. Targeted augmentation of nuclear gene output (TANGO) aims to increase target gene expression by inhibiting nonproductive mRNA splicing events, 38 with recent work demonstrating a successful increase in productive Scn1a transcripts. This was associated with a concurrent reduction in seizure frequency and fatality following ASO-TANGO treatment in a mouse model of Dravet syndrome. 7 A similar method used an oligonucleotide-derived compound (AntagoNAT) to knock-down antisense noncoding RNA that suppresses Scn1a expression. This strategy led to an increase in Scn1a transcript in mice and African green monkeys, and an amelioration of the Scn1a-mediated epileptic phenotype in Dravet mice. 8
Another way to downregulate mRNA levels is using short-harpin RNAs (shRNA) or micro RNAs (miRNA). These tools have shown promise for improving the phenotype of some genetic epilepsies, including dynamin-1 associated epileptic encephalopathy. 15
These therapeutic approaches are very promising, and they have the advantage of targeting mRNA, therefore avoiding expression modulation in cells not expressing the gene of interest. However, one potential limitation is that the transcriptional modulation is not permanent, potentially requiring retreatment in a clinical setting.
Treating the Symptoms
For the overwhelming majority of patients, there is no single identifiable underlying molecular defect at the root of seizures. In cases of refractory focal epilepsy with a defined focus, treating the symptoms (the seizures) is a feasible alternative to resective surgery. Seizures lead to major disruptions in patients’ lives and significantly increase the risk of sudden unexpected death in epilepsy. Gene therapies have a potential role in manipulating the epileptic network and to suppress seizures, usually by either promoting inhibition or suppressing excitation. Many different approaches have been developed so far with some very close to first in-human clinical trials.
Neuropeptides
Neuropeptides are attractive therapeutic targets for their highly specific interactions with target receptors and very low effective concentrations. 39 One of the promising neuropeptides is dynorphin, which interacts with kappa opioid receptors and exerts an inhibitory effect in the limbic system. 40 Adeno-associated viral vector delivered pre-pro-dynorphin in hippocampal regions in rat and mouse chronic models of epilepsy leads to a prolonged suppression of spontaneous seizures, and prevents a decline of learning and memory. 16,17
Neuropeptide Y (NPY) is a secreted neuropeptide widely expressed in the central nervous system, especially in the limbic system, including hippocampus and amygdala. Overexpression of a human NPY in the hippocampus using an AAV has shown an anti-seizure effect (40% reduction in seizure frequency). 18 This effect was most likely mediated by the inhibition of presynaptic glutamate release via NPY receptor Y2 (NPY-Y2). 41 Indeed, the combined overexpression of NPY and NPY-Y2 with a single vector (CG01) showed a more prominent anti-seizure effect. 19
Another neuropeptide implicated in modulating glutamate release is galanin. 42 AAV-mediated overexpression of galanin has shown to decrease evoked seizure threshold. 20
Engineered Channels
The modification of receptors and channels has been used not only to understand basic neurophysiology but also to treat neurological diseases modulating neuronal properties. Both optogenetic activation of inhibitory neurons and optogenetic inhibition of excitatory neurons have been shown to decrease epileptic activity in different rodent models, with several brain regions targeted including cortex, hippocampus, cerebellum, midbrain, and thalamus. 24 -28,31
Clinical translation of optogenetics presents substantial challenges, not least because of the requirement to deliver light to the brain. Although acting with a lower temporal resolution, chemogenetics can also be used for on-demand or controllable modulation of neural circuits. The activity of neurons within the epileptic focus can be controlled, for instance, by using designer receptors exclusively activated by designer drugs. AAV-delivered inhibitory hM4D(Gi) receptors targeted to excitatory neurons, when activated by clozapine metabolite clozapine N-oxide or olanzapine, 43 showed robust anti-seizure effects. 21,22 Recently, a decrease in cortical seizure severity using AAV-hM4D(Gi), coupled with systemic injection of deschloroclozapine, has been shown in NHPs. 23
Another approach to chemogenetics that dispenses with the need for an exogenous ligand to activate the synthetic receptor is to modify the biophysical properties of a receptor of interest to respond to endogenous signals related to seizures. Recently eGluCl (glutamate-gated chloride channel with enhanced glutamate sensitivity) has been shown to be a promising tool to decrease epileptic events in a rat model of neocortical focal epilepsy, using pathological increases of glutamate in the extracellular space as the endogenous activator of the receptor. 29
Endogenous Channels
While exogenous channels could encounter issues with immunogenicity in clinical translation, endogenous channels offer promising alternatives. Reduction of voltage-gated sodium channel Nav1.6 expression, via ASO or shRNA-mediated knockdown of Scn8a, compensates for some epileptic features observed in animal models of a variety of ion channel haploinsufficiencies (Scn1a+/-, Kcna1-/- and Kcnq2fl/fl mutant mice) and temporal lobe epilepsy. 10,34,35 Alternatively, overexpression of the voltage-gated potassium channel Kv1.1 in excitatory neurons in either the neocortex or the hippocampus can suppress spontaneous seizures and epileptiform activity in chronic rodent models of epilepsy. 31,32 Recently, upregulation of Kv1.1 using CRISPRa has also been demonstrated to reduce seizure frequency and improve behavioral comorbidities in a chronic mouse model of focal epilepsy. 30 Another approach to overexpressing an endogenous channel is through activity-dependent promoters that follow neuronal dynamics and selectively target hyperexcitable neurons. When delivered to the epileptic focus of a mouse model of chronic epilepsy using AAV, this tool greatly reduces spontaneous seizures. This approach’s advantage lies in its specificity for pathological neurons both in time and space and its self-regulatory mechanisms. 33
Roadblocks to the Clinic
For treatments addressing a molecular defect underlying epilepsy to be feasible, the cause must be known and identifiable in patients. However, as mentioned above, monogenic epilepsy is rare, and there is a large degree of heterogeneity in the genetic cause of DEEs, necessitating the use of precision medicine to tailor genetic approaches to individual patients. For instance, while ASO and CRISPRa methods for Dravet syndrome work to increase SCN1A expression in the context of LoF-mediated haploinsufficiency, these methods would be contraindicated in a recently reported subset of patients with early infantile epileptic encephalopathy in whom GoF SCN1A mutations have been identified. 44 As precision medicine remains expensive and time-consuming, this introduces significant logistical and financial hurdles before its systematic introduction into the clinic.
Importantly, as these epilepsy syndromes frequently present and progress early in childhood, the question of the most appropriate time of intervention is vital. Genetic interventions may display a temporally narrow window of efficacy, after (or before) which little amelioration of the disease phenotype may be seen. 45,46 This may be especially the case for therapies targeting ion channels expressed only transiently in development—for instance, SCN1A, which peaks at 7 to 9 months of age and decreases in later childhood and adulthood. 47 Further, once secondary sequelae emerge due to the primary pathology, treatment solely of the cause may be inappropriate to address the full syndrome of deficits observed. While some evidence suggests that reestablishing SCN1A expression in older (P30 and P90) Dravet mice reduces spontaneous seizures and rescues some behavioral deficits, 48 it remains unclear how to extrapolate these findings to patients. It is not possible to predict if such interventions later in life will in general successfully rescue the epileptic phenotype, or its sequelae such as developmental delay, at which point further pathology secondary to the epileptic attacks may be permanently established. These encephalopathic features of DEEs may not be entirely secondary to the epileptic features, with some symptoms in Dravet patients emerging independently of seizures. 49 In these cases, it remains unclear if genetic corrections designed to induce seizure freedom will also ameliorate the nonepileptic, encephalopathic features of disease.
In spite of these roadblocks, translation to the clinic is imminent. Indeed, clinical trials for genetic therapies to treat Dravet syndrome and Angelman syndrome have begun. The MONARCH study uses ASO-TANGO technology (STK-001, NCT04740476) to upregulate Nav1.1 expression, with preliminary results suggesting that single and multiple doses of STK-001 are well-tolerated and may reduce seizure frequency. In parallel, with the demonstration of a sufficient safety profile of the transcriptional activator ETX101 in NHPs, 6 the ENDEAVOR trial is a phase I/II clinical trial (NCT05419492) aiming to assess the safety and efficacy of ETX101 in infants and children with Dravet syndrome and is due to begin recruitment this year. Further, a phase I/II clinical trial is ongoing to assess the safety of an intrathecally administered ASO therapy in patients with Angelman syndrome (NCT04259281).
Importantly, treating symptoms remains the only option for many patients without a genetic diagnosis. In practice, it is often difficult to draw clear relationships between genetic landscape and epileptic seizures. Apart from a small proportion of patients with identified familial or de novo monogenic mutations, the majority of patients with epilepsy have complex etiologies with potentially multiple genetic contributions. 50 The high variety of causes of epilepsy, and the likely concurrent high variability of genetic changes in the pathological tissue, complicate the design of any individual genetic targeting tool, especially for epilepsies of nongenetic origin. Even when the genetic cause seems clear, like for Dravet, other genetic variability in patients’ genomes could be pivotal for the development of the pathology. 51 Further, in cases when a pathology with known genetic cause has been consolidated in patients for many years, targeting the seizures could be a promising option. With ongoing research into epilepsy mechanisms, more potential gene therapy targets are being revealed significantly increasing the possibilities for patients. Remarkably, the first gene therapy for epilepsy symptoms using Kv1.1 is due to start this year and is soon recruiting patients with focal epilepsy for a Phase I/IIa clinical trial (NCT04601974). Other approaches are similarly on the path to be registered for their first in-human clinical trials, ushering in a new era for genetic therapies in epilepsy.
Footnotes
Authors’ Note
James S. Street and Yichen Qiu contributed equally.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: GL and YQ are listed as inventors on Patent WO2021191474A; GL has equity in a company that aims to bring epilepsy gene therapy to the clinic, with no involvement with this manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Medical Research Council New Investigator Project Grant MR/S011005/1 and the GOSH/Spark Research Grant V4422 to GL.